Ten Frequently Asked Questions about Serialization

To meet the EU serialization deadline on 9 February 2019, pharmaceutical companies and their contractors have had to reorganize their manufacturing lines and logistics to ensure compliance with the EU’s Falsified Medicines Directive (FMD) of 2011 and the EU Commission Delegated Regulation 2016/161 of 2016. Worldwide, other anticounterfeiting regulations are already in place or coming soon in nations including the US, Saudi Arabia, Korea, and Russia. Thus, drug manufacturers that market products internationally must continue to reorganize their operations to comply with many different standards.
The European serialization scheme establishes specific safety features for prescribed medicines: an antitampering device and a unique identifier. 1, 2, 3, 4, 5, 6, 7, 8 The unique identifier is composed of a unique sequence of a product code, a serial number based on a randomized algorithm, a batch number, and expiry date, and, when required by the local authorities, a reimbursement number. It must be clearly printed on the packaging and encoded using a standardized data structure and syntax in a two-dimensional, machine readable data matrix (i.e., barcode). Before the medicine is dispensed to the patient, the pharmacist at the dispensary or hospital ensures the medicine’s authenticity in an end-to-end verification system by scanning the data matrix so the unique identifier can be compared with the information uploaded in a secured system of repositories. The repositories system for storing serialization data must be set up by the marketing authorization holders (MAHs) and/or the manufacturers (master data and unique identifier commissioning). The unique identifier is then decommissioned when the pack is dispensed within the EU or remarketed to a non-EU country.
To comply with EU serialization regulations, drug manufacturers have been working hard to modernize packaging lines in their production plants with the new printing devices, monitoring systems, and software necessary to serialize drugs subject to prescription and connect labeling information with data repositories at national and European hubs. For the last three years, the ISPE France Affiliate Serialization Workgroup has produced tools for serialization project stakeholders, which can be helpful for new or renewal projects, within the EU or overseas. Some of those tools are highlighted in the following frequently asked questions (FAQs).
Why Serialization Faqs Are Timely
Serialization is one of the biggest information technology (IT) challenges to affect the pharmaceutical sector in the last decade. In a global landscape of constantly evolving regulatory, market, and technical requirements, serialization is, and will likely remain, a complex topic for all industry actors. The Workgroup on Serialization was convened to capture the variety of insights gained by those who have implemented serialization. It has developed this easy-to-understand document to complement (but not replace) regulatory guidance from EMA, FDA, and others with information gathered from the industry. We hope serialization project stakeholders will find these FAQs useful.
Question 1: What Are the Key Components of a Serialization User Requirement Specification?
A user requirement specification (URS) document is the first reference document built when a serialization project is initiated. URS documents are also needed when an existing system must be modified to comply with new regulations.
The URS document specifies all technical and functional requirements to be included in the request for quotation (RFQ) that is sent to all the vendors invited to bid during the sourcing phase of the serialization solution. It includes requirements from all stakeholders of all areas in scope, and there are many stakeholders—serialization affects almost the entire supply chain, extending far beyond the packing hall and manufacturing lines.
When a preferred or preselected vendor is identified before the RFQ, the URS provided with the vendor’s standard commercially available solution could be used. This approach is especially relevant for many of the Level 3 serialization solutions available on the market, which already include standard URS in their documentation. Even if standard URS are available, the user should perform their own risk assessment to ensure that they adequately understand the complexity and novelty of the scope of functionalities to be purchased.
The following list summarizes key components (i.e., chapters) of a serialization URS document. This list is not intended to be exhaustive. If information listed is already available elsewhere, it can be referenced instead of duplicated in the URS. FAQs 2–10 explore the items in this list in greater detail.
- Introduction
- Serialization project presentation and scope (in particular, which regulations must be covered and what kind of track and trace model is involved)
- Operational requirements
- Constraints on solution specification and operations
- Requirements for the serialization solution throughout its life cycle
- Approvals
Question 2: What Should Be Emphasized in the Presentation of the Serialization Project?
This project presentation section describes the overall purpose and scope of the serialization project—specifically, the objectives and deliverables that have been assigned to the project and the implementation strategy. For example, is the objective to meet only European regulatory requirements, or is the company looking for a solution that can accommodate more complex functionalities and/or the next wave of requirements to be published? Does the company hope to enhance its return on investment by implementing functionalities that will bring added value to its products and services?
Along with regulatory compliance (which is a must-have requirement), serialization projects may have additional benefits, such as the following:
- Tools to identify and help investigate problems: For example, the time stamp of the serial number operation on the packaging box can be useful when investigating complaints about products.
- Tools to help track and trace stock: The serial number can also be used to track stock in the supply chain from the manufacturer to the final patient, as well as to trace products returned to any part of the supply chain.
The presentation section should clearly identify the project scope by answering the following questions:
- Is the project limited to production site(s)? How many sites are in the scope? Does the project include sites other than manufacturing sites, such as general offices?
- How many packing lines are to be equipped with serialization?
- How many product families and SKUs are in the scope?
- What is the packaging configuration of the saleable unit?
- Does the management of serial numbers (creation, reporting, etc.) need to be included in the project?
- How many interfaces are in the scope? (For example, does the serialization solution need to interface with ERP (enterprise resource planning) or MES (manufacturing execution system) or support reporting to local or regional regulatory agencies?)
- Is there any need to connect the serialization solution to partners (e.g., joint ventures, alliances) or specific vendors?
- Which processes are in the scope? What are the applicable regulations for those processes?
- Is aggregation to be included if the company is supplying non-EU markets from Europe? If so, is aggregation to be done at just the case level, or will it be extended to pallets for the larger volumes and for efficiency purposes?
Question 3: What Are the Operational Requirements?
The following processes will have to be extensively specified to ensure generation and management of data in accordance with all regulatory requirements:
- Functions performed during the serialization process (see FAQ 5)
- Definition of data and data quality (see FAQs 6 and 7)
- Data management
- Technical requirements (see FAQ 8)
- Interfaces (see FAQ 9)
- Environment and corresponding installation prerequisites
Process descriptions and documentation (e.g., flowcharts) can be included as relevant in any of these sections of the URS.
All requirements specified should be pragmatically achievable and verifiable, although these objectives may be difficult to specifically define and subject to multiple interpretations. A good approach is to define criteria to measure how requirements are achieved. These criteria can be used to evaluate whether vendors that participate in the RFQ process provide proposals that reflect the business need as closely as possible. Additionally, these criteria could be used as benchmarks during the implementation phase.
Question 4: Which Functions Are Typically Performed during the Serialization Process?
The following functional requirements must be specified and documented in the URS:
- Calculations: All critical algorithms, such as those that ensure compliance with regulatory or key internal requirements, must be duly described. The supporting scientific sources and calculations for each algorithm must be documented, and the relevance of these sources and calculations for each critical algorithm that has been adapted for the scope of the serialization project must be clearly articulated.
- Performance of randomization algorithms: Randomization shall be performed within a reasonable time frame. This performance level will need to be maintained across the life span of the project, in accordance with not only current but also future business needs of the company.
- Volume represented by codes generated: The number range that shall be dedicated to serial numbers will need to significantly exceed the number of serial numbers expected to be consumed across the life span of the serialization project (consider a minimum life span of 5 years according to Delegated Regulation 2016/161 Article 4 d). Each serial code generated will be controlled to ensure its unicity in the considered product code. If the number range for serial numbers is too short, it will become increasing difficult for the randomization algorithm to generate new serial numbers over time. Having an expansive range of serial numbers will prevent the randomization processing time from exponentially increasing, which could cause serialization systems to collapse.
- Collision rate: This is the risk that the same serial number will be generated twice. Best practices recommend that a system limit this risk to less than 1 chance out of 10 million. To guarantee an acceptable collision rate, the ratio of the number of all serial numbers generated to the serial number range must remain below 1:10 across the serialization project’s life span.
- Control of serial codes unicity before a batch of serial codes is released to manufacturing: This activity is the responsibility of the function generating all serial codes. The process to perform this activity will need to be specified.
- Reconciliation of serial codes generated at the end of manufacturing: The process to perform this activity will need to be specified. For the EU market, only serial numbers of saleable products are communicated to the central database. Serial numbers of retention samples and stability samples are recorded for internal follow-up, but they are not communicated to the central database. For security reasons, it is crucial that all serial numbers that have been uploaded on the packaging line are not reused for any other production.
- Security: Measures to control access to serial codes and protect data must be specified.
- Audit trails for all users and activities, including all changes performed: Processes to support audits must be explained.
- Electron signatures: These must be defined and their usage specified for every critical activity that will be performed in the system, such as serial number generation or batch certification. For the EU market, batch certification is performed by the qualified person (QP) responsible for ensuring that each individual batch has been manufactured and checked in compliance with laws in force in the member state where certification takes place, in accordance with the requirements of the marketing authorization (MA) and with Good Manufacturing Practice (GMP). After certification by the QP, the product is released for sale or supply to the market (according to Delegated Regulation 2016/161 Article 33 1, the MAH shall ensure that the unique identifier is uploaded to the repositories system before the release for sale or distribution by the manufacturer).
- Reporting: Every aspect of reporting (e.g., manufacturing reports, reconciliation, errors) must be covered in the URS document.
- Error messages: Specifications must ensure that error messages generated by the system will be clear and unambiguous.
Question 5: What Does Data Quality Mean for Serialization Data?
Data quality can be expressed through six principles:
- Accuracy: Data can be considered accurate and not altered by any event or whenever it is handled. Their definition remains consistent across the information processing chain and thus in all systems where they are handled. All modifications receive a time stamp (hence the importance of time-stamped audit trails).
- Completeness: Data are constantly available and comply with standards and controls in place. No data are lost after a transfer from one system to another.
- Availability: Data are readily available and can be handled directly without additional transformation. There is no need to manipulate data files; all systems use the same standard data format.
- Integrity: The data attributes are consistent in all systems where the data are handled. Each value has a fixed definition and is decoded and used consistently by all users.
- Secured data sources: Only authorized and validated data sources are used. All exchanges between systems are secured so that data integrity and availability can be ensured at all times.
- Fit for use: Data meet all needs of all stakeholders and allow the company to comply with all regulations enforced in the project scope.
Question 6: What Data Need to Be Secured and According to Which Principles?
The Delegated Regulation R2016/161 and European Medicines Verification Organisation (EMVO) guide identify 18 types of data that shall be managed and exchanged in a secured way. Protection of the data needs to be ensured throughout all data manipulation and transformation steps.
The following “ALCOA+” principles for the scope of data integrity can be used as well for data security. ALCOA+ data are:
- Attributable: The roles in the organization that are entitled to access and manipulate serialized data are clearly defined.
- Legible: Unauthorized recreation of serialized data without an audit trail is not allowed.
- Contemporaneous: Data are created and time stamped in real time, when the serialization takes place, not after the fact.
- Original: Original data, or a secured copy, can be accessed.
- Accurate: It is possible to confirm data accuracy against a known specification.
- Complete: The completeness and consistency of combination of a global trade item number (GTIN) with serial numbers, including their attributes, must be maintained at all times. To ensure complete data, changes made to the data and deviations identified along with their resolution must be taken into account.
- Enduring: Data storage and access exist for the entire life cycle of all serialization data.
- Available: Data are available for review at any time throughout their entire life cycle.
Question 7: What Technical Requirements Must Be Defined?
All technical requirements of a serialization information system shall be defined, including the following:
- Processes for managing changes to system operations: How will changes that affect the way the system operates, such as start-up and shutdown sequences, test sequences, and cut-over phases, be managed?
- The disaster recovery process: The system must include a pragmatic process that can reconcile all materialized data (already printed on product packs) vs. all serial numbers that not yet been fully processed. The system must be able to generate disaster recovery reports, including how to take into account master data and variable data on hand. The disaster recovery process must also be capable of restoring a running production environment within a minimal time frame (the maximum allowed time to restore a live and functional production environment shall be part of the specifications documented).
- The required level of performance and expectations for response times: These must be expressed with clear values, and without any ambiguity. Level-of-performance specifications must cover what will be done to maintain data consistency in case of system breakdown, and the reconciliation processes. Response-time specifications can influence equipment choices, based on how the serialized solution will be operated. For example, if the check of unicity of the serial number will be done in real time during manufacturing or product picking, one will need response times of about a millisecond from the data server on each query. One way to handle this kind of requirement during packing could be to host the range of serial numbers manipulated on the server of the manufacturing line during production.
- Data storage: How will data be stored? What will be the maximum allowed amount of data stored?
- Updates: What will be the process for system updates and system patches?
- Specific equipment/system requirements: These can include requirements for onboard devices, especially for logistics-related activities on serialized products and requirements related to systems efficiency (loading times, screen refresh times, time to generate reports, etc.). The actual demonstrated performances of all user interfaces will be critical to ensure fluid operation of the serialization system and effective manipulation of serialized serialized data, and, ultimately, to maintain trust and adherence to processes from the operations teams. When aggregation is implemented, a typical operation such as scanning serial numbers on product cartons or serial number of cartons on a pallet must be done with maximum fluidity to be integrated seamlessly into standard operating procedures of the warehouse operators.
- Adaptability: This is a major requirement. Because regulations and market requirements are constantly changing, the serialization system must have the critical capability to seamlessly adapt the database to accommodate new data elements or changes in data structure. The more markets are included in scope, the more critical it is that the system meet this technical requirement.
- Security: This is a critical function to be managed by any electronic or IT system. All security-related requirements about system access, networks, firewalls, VPN, and so on must be included.
Question 8: What Interfaces Must Be Specified?
Specifications for interfaces between systems should be classified as follows:
- User interfaces: These should be tailored to all authorized personnel who will have access to and play a role into the serialization process (e.g., operators, team managers, systems administrators).
- Interfaces with other systems: Having the capacity to connect the serialization solution to a variety of both internal and third-party systems is one of the key challenges of serialization. Examples of interfaces to include in the URS might include:
- Interfaces between a marketing authorization holder (MAH) and its contract manufacturing organizations (CMOs)
- Interface between manufacturing and distribution centers
- Interface between Level 4 serialization solutions and ERP or MES
- Connections between all actors in a supply chain
- Connection to a warehouse management system
- Connection with regulatory hubs
- Connection with distributors
This list is only a partial one. It generally identifies interfaces that involve several actors, which may be using various, very different types of interface technology and information systems. Thus, the interface system may need to meet a variety of different needs and address many local constraints. There are also several ways to implement Electronic Data Interchange (EDI) and Electronic Product Code Information Services (EPCIS) standards.
It is important to keep in mind that following a standard does not necessary mean following a unique format for communications. There are many different ways to connect several systems, using several formats of data, and the appropriate approach can depend on local constraints. For example, requirements for the EU are not the same as those for the US, China, Brazil, or South Korea, and some EU-member states may have requirements that are more stringent than the EU Falsified Medicines Directive. Sometimes, market-driven requirements may be more stringent than regulatory ones (as in the case of the US market). Also, third-party logistics providers may use processes that are not yet covered by current regulations, such as aggregation for some markets.
The following are some of the distribution-related transactions that need to be supported by a serialization solution:
- Goods receipts (including batch, serialized, or aggregated data)
- Where-used and inventory reconciliation reports
- Proceeding to shipments
- Product returns and the batch recall process
- Support of investigations required by a customer or partner
The referent used by each actor is usually based on the GS1 standards such as main distribution-related standards or EDI/EPCIS communications. While EDI/EPCIS protocols use a common standard, communication formats may vary from one partner to another. These standards are complicated and have been in place in the supply chains for some time already; hence, it is preferable to consider adapting the serialization system to them vs. trying to modify standards or protocols to fit the system. Other factors that may encourage users to adopt a flexible serialization solution are the many proprietary communication protocols on the market as well initiatives such as Open Serialization Communication Standard (OPEN-SCS), which aim to establish technical communication standard.
Question 9: What Are the Environment and Corresponding Installation Prerequisites?
The definition of the environment in which the serialization solution will operate should include the following items:
- Footprint: The location where the serialization solution is implemented or the configuration of that location can affect the solution’s performance. Factors such as long-distance connections or a relatively small data center are examples of footprint-related concerns.
- Environmental factors: Considerations to specify could include temperature, moisture, interferences, presence of perturbating sources such as external high-frequency signals, ultraviolet sources, dirt, high-powered vibrating systems, electromagnetism sources, or the proximity of a sterile room.
- Access control and all security procedures: These prerequisites must be clearly defined.
- Energy requirements: It is important so specify voltage and frequency, waveform, protection against voltage drops, and other energy-related matters.
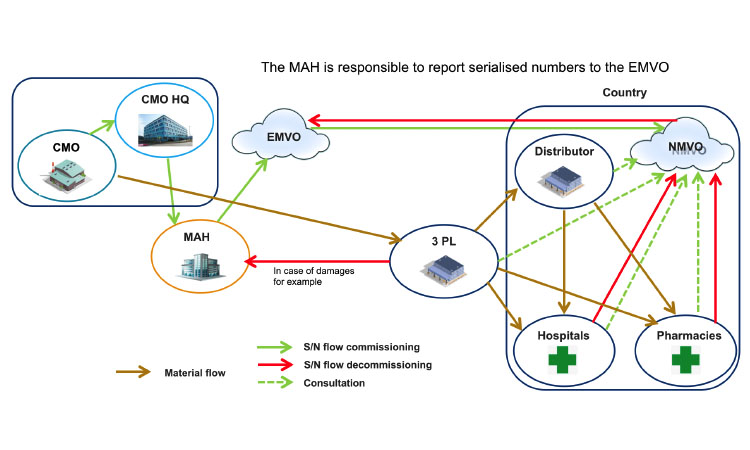
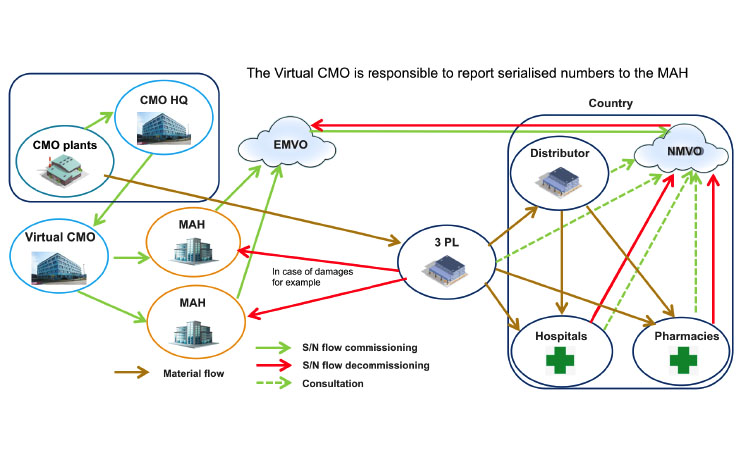
Question 10: What Are the Possible Business Cases Where Cmos Are Involved in Serialization?
Two main business cases can be considered: between an MAH and its CMO (Figure 1) or between an MAH and a virtual CMO (a CMO that contracts manufacturing to other service providers) (Figure 2). Many variations in these business cases have been identified by the Workgroup and will be discussed in later versions of this document.
The Future of These Faqs
The ISPE France Affiliate is hosting the Serialization Workgroup that has developed these FAQs, but the Affiliate cannot be held responsible for any statement or recommendation contained in this guide. We encourage the readers to contact the Workgroup with new questions, thoughts on points that seem unclear, and other feedback. Part of the Workgroup’s mission statement (and what we also find exciting) is to respond to your feedback. This guide will be regularly updated to reflect the issues you raise. Contact the ISPE France Serialization Workgroup.


