Breaking with Tradition: Laying the Foundation for Validation 4.0
A fundamental GMP requirement is that processes, systems, and methods used to produce medicines and treatments are validated, meaning their fitness for a purpose is demonstrated. If Industry 4.0 is to succeed in the pharma space as Pharma 4.0™, we need new paradigms for validation across the value chain that use new technologies to improve product quality and the safety of medicines and treatments for the patient.1 We must transition from our old ways of approaching compliance to embrace this new age of data-powered technology. This article lays the foundation for shifting our mindset and achieving Validation 4.0.
Despite notable initiatives since the early 2000s from regulatory leaders such as ICH, the US FDA, and EMA, the pharma industry has struggled to move on from legacy quality management approaches. The original ICH statements that led to Q82 were intended to modernize pharmaceutical quality systems through a focus on quality risk management3 and quality by design (QbD). These concepts should have also been a basis for implementing new technologies, as the ICH concepts offer a framework to cope with inherent variability, and allow for a compliant flexibility needed for manufacturing increasingly sophisticated, even personalized medicinal products. However, for pharma organizations and regulators alike, the topic of validation has been a central obstacle to adopting new concepts for quality.
As we look to overcome this obstacle, several questions about validation must be addressed: What would it mean to not write documents in the regulated environment? What do QbD and data integrity mean when applied to a manufacturing facility? Can we move beyond three stages of process validation1 to defining a control strategy and then continuously verifying that control? We invite you to think about what the answers to these questions might mean for you.
Rethinking What We Do
To illustrate the need for a new mindset, let’s start with a simple example derived from the definition of validation: establishing documented evidence of fitness for purpose. Generally, this principle has been interpreted as a requirement for documentation in either physical or electronic form. For example, during up-front validation testing, documents are used to record how the introduction of a process or system was controlled to ensure it operates correctly.
However, for Validation 4.0, we need to move on from creating historical documents of what was tested to focus instead on real-time verification of product quality by managing specification and evidence data around a process that is in a state of control throughout the life cycle. Standalone documents are clearly not suited to continuous verification, and the masses of documentation created by both suppliers and regulated companies in the name of validation are inefficient, difficult to maintain, and perhaps not auditable.
In the Industry 4.0 world, connected data and systems are subject to rapid and constant change for iterative improvement, and they are now being enabled for autonomous improvement. Instead of relying on difficult to maintain silos of documentation, we look toward digital artifacts managed with appropriate tools that can instantaneously provide reporting and notifications on the state of control. The systems used widely today by agile software developers for multitenant cloud solution providers are a good reference point for Validation 4.0: by adopting the usual tools of software engineers, we can leverage and integrate quality management efforts into our ongoing activities of continuous verification beyond what is possible with static documented evidence.
Data Is the Foundation
Data integrity has been a buzz term for years now. A whole sub-industry has been built around this concept, and yet we still fail to truly embrace what it means and how to implement it. Data is the foundational element of validation and the basis for decision-making. When we consider validation, we need to shift our focus to how we control the data that allows us to make GxP decisions, and look at validation under a QbD lens.
Validation 4.0 is a holistic and integrated approach to validation with process and data flows at its foundation. Let’s follow the product journey that starts with an unmet patient need. To characterize a product intended to meet that need, QbD principles suggest that we begin by defining critical process parameters (CPPs) and critical quality attributes (CQAs). Critical process parameters are defined to control the quality outputs, and we have to understand the critical quality, process, equipment, and material attributes to measure and control them within a defined range of variance that produces a quality product. This design space is an output of product development and the basis for handling inherent variability. It is then expected that as the product moves to actual production, real-time process data will be available and monitored so that the design can be refined. By bringing the QbD concept into Validation 4.0, we further encourage the early definition and use of data points to control and ensure the desired product quality attributes.
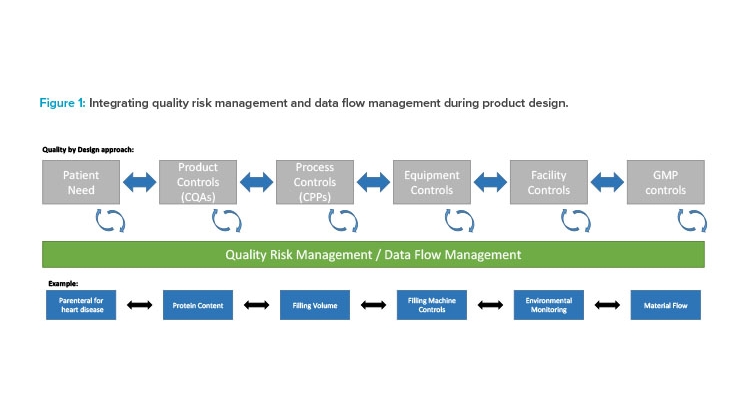
A quality risk management approach should be integrated with QbD and applied at the process and data flow level as a part of the design process (Figure 1). First, traditional user requirement specification (URS) statements are replaced by process and data maps (PDM). In Industry 4.0, data are referenceable, used throughout a process, and a basis for making effective decisions. By building a validation model that incorporates the process and key data early in design, we get a head start on defining the associated risks and the needed controls.
Following the initial process and data definition, the focus of validation changes from qualification testing to ongoing and constant assurance that the needed controls are in place and operating correctly. This continuous verification of the process and risk is the primary evidence that the process is in a state of control. By using real-world data to feedback into our process, data, and risk evaluation, we can be assured that our products are constantly manufactured and released based on sound data, and through this model, we can continuously reassess risk conditions and handle inherent process variability.
From Process Validation to Validation 4.0
As the US FDA has stated, “effective process validation contributes significantly to assuring drug quality.” 4 Process validation is a series of activities that occur over the life cycle of the product. Table 1 presents how the three stages of process validation can apply to Validation 4.0.
Figure 2 illustrates the holistic view of the Validation 4.0 life-cycle model and how it relates to the product life cycle.
| Stage | Process Validation | Validation 4.0 | Justification for Validation 4.0 |
|---|---|---|---|
| 1 | Process design: • Build and capture process knowledge and process understanding. • Create quality target product profile. • Identify CQAs. • Design a process suitable for routine commercial manufacturing that can consistently deliver a product meeting the CQAs. • Define CPPs. • Establish a control strategy for ensuring the product will meet the quality requirements. • Conduct risk assessment(s). |
Holistic planning and design: • Define requirements as process and data flow maps • Identify critical attributes and parameters from the process, materials, equipment, and external threats for ongoing monitoring. • Conduct process and data flow risk assessment at the design stage, incorporating criticality and vulnerability to define the control strategy, and to implement data integrity as a fundamental aspect of QbD. |
• The output of this stage is a holistic control strategy that builds on knowledge from product development and focuses mitigation and decisions on the data. • Data-centric approach provides a basis for utilizing systemized tools for process design and enabling digital twins. |
| 2 | Process qualification/process validation: • Collect and evaluate data on all aspects of the process (from raw materials to finished and packaged materials, as well as the facility). • 2.1: Qualify facility and equipment (URS, factory and site acceptance testing, and design, installation, operational, and performance qualification). This may involve specific equipment qualification considerations. • 2.2: Evaluate the process designed in stage 1 to verify that it can reproduce consistent and reliable levels of quality. |
Verification of controls: • Verify with evidence that the controls defined in the process and data flow risk assessment are in place and operate appropriately to provide the expected risk mitigation. • Automate to rapidly ensure that planned controls are in place/effective. |
• Through model maturity and systemized tools, stage 2 may no longer be required because design includes the controls needed to ensure a product is fit for use; instead, can move directly to continuous verification. • Process results, risk controls, and parameter verification are moved to stage 3, where monitoring of process conditions and dynamic risk aspects and real-time data determines whether the process and controls are operating correctly. |
| 3 | Continued (ongoing) process verification: • Verify in an ongoing manner that the process continues to deliver consistent quality. • Detect and resolve process drift. |
Continuous verification of implemented controls: • Link the operational-stage data back to the process and data maps and specifications to verify actual risk/ control status continuously. • Incorporate data from across the value chain (from raw material suppliers to patients) and product life cycle to evolve the control strategy into a holistic control strategy. |
• Handling inherent variability and change in process and risks is part of the overall method. • Real-time data are used to review whether initial assessments remain valid and to determine controls that enable flexibility in manufacturing (e.g., process analytical technology). • Through model maturity, opportunities are provided to apply digital twins,* analytics, and machine learning to simulate the impact of proposed changes before they are operationalized. |
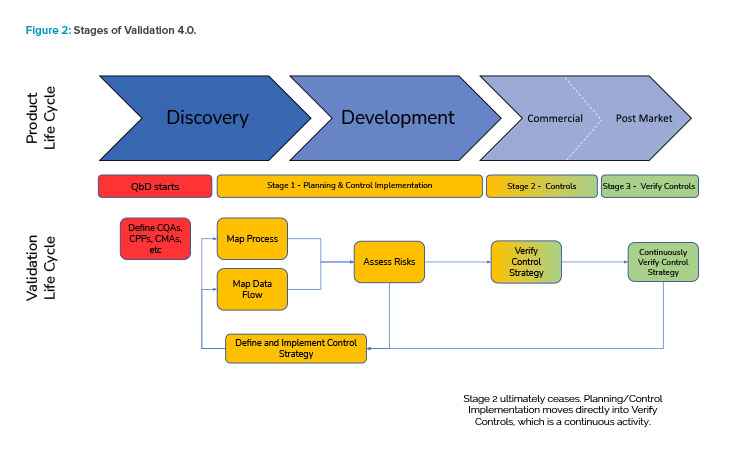
In this model, data from across the product life cycle are key in creating holistic control strategies and continuously verifying them to demonstrate fitness for use, and real-time data from the actual operating process and its critical parameters are checked as part of continuous verification. Additionally, ongoing assessment of pro-cess/data risks is part of the overall integrated method to understand whether a process is in control, to evaluate inherent variabilities and refine control limits, and to consider dynamic risks as they change and trend over time.
Conclusion
By moving to a process- and data-centric approach to validation, and finally establishing a baseline for incorporating QbD, the pharma industry can move to continuous assurance of product quality throughout the product’s life cycle, and at every point in time. The Pharma 4.0™ Special Interest Group recognizes that we do not yet have all the answers, but it is imperative that industry stakeholders work together now to ensure validation moves forward to take full advantage of the positive disruption of digitization, which may include automation, process analytical technology, and feedback loops, as well as data conversion and accumulation. The Validation 4.0 working group is currently working on next steps, including case studies. Please reach out to our working group via the authors with your comments, feedback, and any examples to help shape Validation 4.0.




