A Vision for ICH Q12: Current Experience, Future Perspectives
Management of global postapproval chemistry, manufacturing, and controls (CMC) changes is a growing challenge for industry with many issues. ICH Q12 (Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management) is a transformative document shaping global regulatory postapproval submissions that will help alleviate some of these issues.
Regional regulations necessitate the development and maintenance of country-specific regulatory documents that contain different quality information for the same product to meet individual country requirements. In some instances, simultaneous operation of different manufacturing processes or redundant testing may also be needed for the same product to ensure its uninterrupted availability for patients in various global markets. Increased inventory segregation and the potential for errors in manufacturing and regulatory compliance as well as varied submission, review, and implementation timeframes add to the complexity in the oversight of global commercial product supply chains. Thus, different regulatory requirements around the world are currently a disincentive to making innovative changes or improvements to increase process efficiency and robustness.
The ICH Q121 guidance provides a framework to facilitate the management of CMC changes in a more predictable and efficient manner, building on the science- and risk-based approaches outlined in the prior ICH Quality Guidelines—ICH Q8(R2), Q9, Q10, and Q11. A well-established and effective company pharmaceutical quality system (PQS), in compliance with regional GMP requirements,2 is critical to achieve the full potential of the ICH Q12 concepts.
Postapproval changes can be categorized using risk-based principles and reported to regulatory authorities based on standardized terminology (prior approval, notification-moderate, notification-low) or not reported (i.e., documented within a company’s PQS). Thus, increased product and process knowledge can contribute to a better understanding of which post-approval changes require a regulatory submission. Other key regulatory tools de-scribed in ICH Q12 include the following concepts:
- Established conditions (ECs): Legally binding information considered necessary to assure product quality. Changes to established conditions require regulatory reporting according to the above standardized terminology. Supporting information is still required to accompany established conditions.
- Postapproval change management protocols (PACMPs): These protocols provide predictability regarding the information required to support a change and the type of regulatory submission based on prior agreement.
- Product life-cycle management (PLCM) document: Central repository for established conditions, reporting category for making changes to approved established conditions, PACMPs (when proposed), and any post-approval CMC commitments.
This article provides a description of the current regulatory environment and status of ICH Q12 implementation at the time of writing. It also provides case studies from an industry perspective that serve as practical examples of how industry has interpreted ICH Q12 and has applied ICH Q12 tools to improve PLCM.
| Country/Agency | Current ICH Q12 Implementation Status* |
Unique Considerations/Challenges | Reference |
|---|---|---|---|
| US FDA | ICH Q12 and Implementation Considerations Guidance published on the FDA website. The 2015 draft guidance on established conditions was withdrawn. | Overall, the concept of established condition is consistent with FDA regulations in 21 CFR 71 314.70(a)(1)(i), 314.97(a), and 601.12(a)(1). |
5, 6 |
| EU EMA | Note on ICH Q12 implementation guidance issued |
Certain ICH Q12 elements such as established conditions and PLCM documents are not compatible with the current legal framework. Recently announced EU legislation revision initiative may change this and enable the use of all ICH Q12 tools. | 7, 8 |
| Japan PMDA | In progress | Established condition and PLCM documents will need to be aligned with the Japanese Application Form. PACMP is a novel concept that requires the revision of existing legal framework (ongoing). | 9, 10 |
| Canada (Health Canada) | In progress | Established condition and PLCM will need to be aligned with the Canadian Certified Product Information Document (CPID). There has been limited experience with PACMPs so far. |
11 |
| Brazil ANVISA, China NMPA, Korea MFDS, Singapore HSA, Switzerland Swiss Medic, Chinese Taipei TFDA, Turkey TITCK | In progress | Many countries have initiated pilots and consultations in order to get more insights into ICH Q12 concepts, define implementation approaches, and revise the underlying legal frameworks. |
3 |
Current Adoption by Global Regulators
The final draft of ICH Q12 was adopted by the ICH Assembly in November 2019 (Step 4), and the guideline is now in Step 5, with regional implementation efforts in progress as summarized in Table 1.3 The ICH Implementation Working Group (IWG) was established to develop a comprehensive training program to “facilitate an aligned interpretation and a harmonized implementation of ICH Q12 in ICH and non-ICH regions.” 3 According to the ICH website, the current ICH Q12 IWG work plan foresees the completion of training program preparation later this year, followed by the initiation of training activities in late 2021.4
The European Medicines Agency (EMA) was one of the first global health authorities to adopt ICH Q12 in January 2020 and to issue its implementation guidance in March 2020.8 Despite the extensive discussions and negotiations during ICH Q12 development and endorsement, the incompatibilities between certain concepts in ICH Q12 and the existing EU legal framework (e.g., the Variations Regulation (established condition) No 1234/2008) could not be fully resolved, preventing the full implementation of ICH Q12 in the EU.
EMA’s implementation guidance emphasizes that “the legal framework always takes precedence over technical and scientific guidelines”,7 which means that one must always default to the requirements laid down in the current EU Variations Regulation and associated EU Variations Guidelines. As such, scientific risk-based approaches to defining established conditions and associated reporting categories, as described in Chapter 3.2.3, and the PLCM document, as described in Chapter 5 of ICH Q12, cannot be currently applied in the EU until the legal framework is revised. However, other ICH Q12 tools and enablers, such as quality risk-based postapproval change reporting categories using PACMPs, are considered compatible and ready for implementation. Recently, it was announced that the European Commission has launched the process for revision of the EU’s pharmaceutical legislation, issuing a “combined evaluation roadmap/inception impact assessment.”7 One of the stated goals of this revamp includes an attempt to address the “inefficiency and administrative burden of regulatory procedures” in order to achieve the “simplification and streamlining of procedures and internal processes to reduce timelines and regulatory burden.”7
The US FDA published the final ICH Q12 guideline and annexes on its website in May 2021.5 This guidance is replacing the draft 2015 FDA Guidance on Established Conditions, which accordingly has been removed from the FDA website. In addition, the FDA has issued a draft guidance with specific considerations around the ICH Q12 implementation, which complements ICH Q12 and clarifies “how the ICH Q12 tools and enablers can be implemented within the US regulatory system.”6 The guidance reflects key lessons learned from the 2019 FDA Pilot on Established Conditions,12 providing detailed recommendations on how to define, submit, and maintain the proposed established conditions. It also clarifies the relationship between ICH Q12 PACMPs and FDA comparability protocols, explains how to translate ICH Q12 postapproval change reporting categories to the existing FDA supplement categories, and illustrates the use of a PLCM document with specific examples.

In Japan, the PMDA has established an internal working group to facilitate the implementation of ICH Q12. One of the interesting aspects that will need to be addressed is the relationship and any opportunities for alignment between the Japanese Module 1 Application Form that contains the “ap-proved matters” and the ICH Q12 concepts of established conditions with associated reporting categories and PLCM document. Regarding PACMPs, there is currently no similar concept in Japan and therefore, a change in the national regulations will be required to implement it. Japan has taken a proactive approach to gaining experience with PACMP, with MHLW/PMDA starting a pilot program for PACMP in April 2018 prior to ICH Q12 reaching Step 4.10 A PACMP mockup that reflected the contents of the Application Form for Japan was created.9 Following the pilot, the PACMP was introduced into the regulation as the “Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices” and will come into effect in 2021.
Health Canada has announced its target timeframe for implementation of ICH Q12 to the second half of 2021 to “allow sufficient time for the preparation of regulators and stakeholders.”11 Toward that goal, Health Canada planned to launch stakeholder consultations in 2021 to gather feedback on the final elements of the implementation of the Q12 guidance in Canada. Similarly, in Japan there are some opportunities for alignment of the Canadian CPID with the ICH Q12 concepts of established conditions and PLCM document, as well as more widespread acceptance of PACMPs.
Industry Perspectives
The stage of ICH Q12 implementation varies across both global health authorities and industry. Industry engagement with regulators on adoption of ICH Q12 concepts and the extent of internal adoption of ICH Q12 concepts within an individual company’s own PQS framework have shaped industry experience and perspectives. Several companies participated in the FDA’s Established Conditions Pilot Program as an opportunity to engage with the FDA in defining and proposing established conditions. The pilot program provided the FDA with an opportunity to engage with sponsors and gain practical experience in (a) as-sessing proposed established conditions, (b) engaging with applicants during the review to refine proposed established conditions, (c) ensuring assessment decisions can be made without impacting user fee timelines, and (d) identifying agreed-upon established conditions at time of approval.12 Experiences with the pilot program varied widely across industry participants, and sponsor perspectives are summarized below where applicable.
Amgen
Amgen focused on a drug product container closure change for a biologic molecule as part of the FDA established conditions Pilot Program. Amgen used an enhanced approach as described in ICH Q12 to propose established conditions and corresponding reporting categories in a PLCM document. The enhanced approach was based on an understanding of the interaction between process inputs and quality attributes of the molecule. Proposed reporting categories for established conditions were based on process and product knowledge, as well as Amgen’s established PQS utilizing a risk-based approach.
Proposed reporting categories for established conditions were based on (a) potential risk to product quality, (b) experience in changing the proposed established condition, and (c) capability of PQS in managing changes and an effective continued process verification (CPV) to provide ongoing assurance the process remains in a state of control. Amgen’s PQS was defined in the PLCM document to address several key quality process elements, including those necessary for ICH Q12 implementation. Amgen’s PQS can appropriately identify and mitigate risk, develop robust plans for change implementation, and can effectively assess impact to product quality (Figure 1).
The initial scope of the change included a drug product primary container closure change with no modification to drug product formulation. Established conditions and corresponding reporting categories were therefore assessed for the relevant drug product sections of the dossier. A team of cross-functional subject matter experts (SMEs) assessed established conditions, which were filed as a PLCM document in 3.2.R (regional section of Module 3). For changes with any potential risk to product quality, Amgen assigned established conditions and associated reporting categories in a tiered approach based on risk severity. Non-established conditions were also described in the PLCM document to define supportive information that would solely be managed by the PQS. Amgen is actively pursuing engagement with FDA and other health authorities to realize the full potential benefit of ICH Q12.
Janssen
Janssen’s initial approach around the implementation of ICH Q12 has focused on manufacturing process descriptions for conventional small molecule solid oral dosage forms. As the global health authority expectations around the level of detail provided for process parameters (critical and noncritical) in manufacturing process descriptions continue to increase,13 Janssen has sought to leverage key ICH Q12 enablers such as established conditions to mitigate the increase in the level of process parameter details in regulatory filings and reduce the potential postapproval change reporting burden.
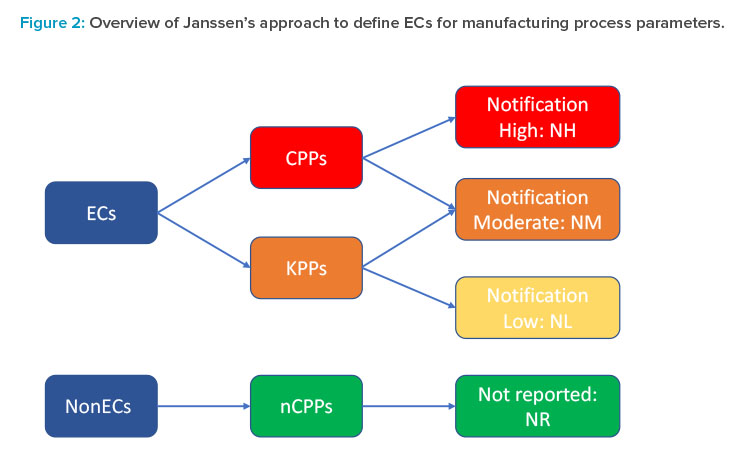
To achieve that goal, the first step was to develop a more granular risk assessment and filtering tool that would permit moving beyond the current binary critical process parameter (CPP)/noncritical process parameter (nCPP) paradigm and provide a clear framework for determination of established conditions and associated risk-based reporting categories for all process parameters using the enhanced approach. A third category of key process parameter (KPP) was introduced for internal use only and defined by Janssen as parameters of the manufacturing process that may have a relationship to a CQA but have a reduced risk of impacting the safety or efficacy of the product compared to a CPP. Each functional area could then further refine and elaborate this general definition in terms of what “reduced risk” means in practice in their area, as well as to propose further risk filtering and ranking of CPPs and KPPs based on the degree of their impact on CQAs and the overall control strategy.
For example, for drug substance synthesis, process parameters impacting an impurity observed at intermediate stages but not in the final drug substance can be considered as KPPs. For the drug product manufacture, in cases where the primary CQA control strategy is focused on unit operation out-puts (e.g., in-process controls [IPCs] or output CPPs), the input process parameters that directly influence these outputs may be defined as KPPs. Finally, each risk-based category of process parameters was mapped to the appropriate postapproval reporting category (Figure 2). It is important to note that not all process parameters in the same criticality category (e.g., CPPs) are necessarily assigned to the same reporting category (e.g., notification-high), as this assignment depends on the actual risk to quality and can depend on the nature and directionality of the change (e.g., expanding versus tightening of control ranges), the ability to control a given parameter within its operating range, and the overall control strategy (e.g., the presence of other down-stream control elements). Janssen is currently piloting this approach in select markets and plans to refine and adjust it based on the feedback received.
GSK
GSK’s preparation with regard to ICH Q12 includes a global cross-modality team supporting adoption and implementation of ICH Q12 principles and tools by leveraging current quality by design (QbD) practices and a robust PQS. Participation in the FDA established conditions Pilot Program provided an opportunity to gain feedback from the agency regarding GSK’s strategy for identification of manufacturing-process-related established conditions. GSK submitted a prior approval supplement (PAS) to support a column-size diameter change for a chromatography unit operation of a mAb. The science- and risk-based approach utilized by GSK was based on the failure modes effects analysis (FMEA) technical risk assessment evaluating criticality of the parameters and controls. Established condition identification was based on the severity scoring regarding the impact to CQAs. The strategy utilized a continuum approach where (a) a high severity score (i.e., extremely severe, moderately severe) was identified as an established condition, (b) a low score (i.e., not severe) was defined as a non-established condition, and (c) a medium score (i.e., slightly severe) required additional assessments to determine if it was an established condition or non-established condition. Further assessments for slightly severe were based on product and process knowledge (i.e., process characterization data). If process characterization data demonstrated a parameter or control did not have a practical impact on a CQA over a reasonable range, it was considered a nonestablished condition. However, if knowledge and data were not available to assess the impact, it was classified as an established condition until further information would be available. Following the established condition identification exercise based on the continuum approach, 21 registered criteria were reduced to seven criteria defined as established conditions, whereas the remaining 14 criteria were defined as nonestablished conditions to be managed under the PQS.
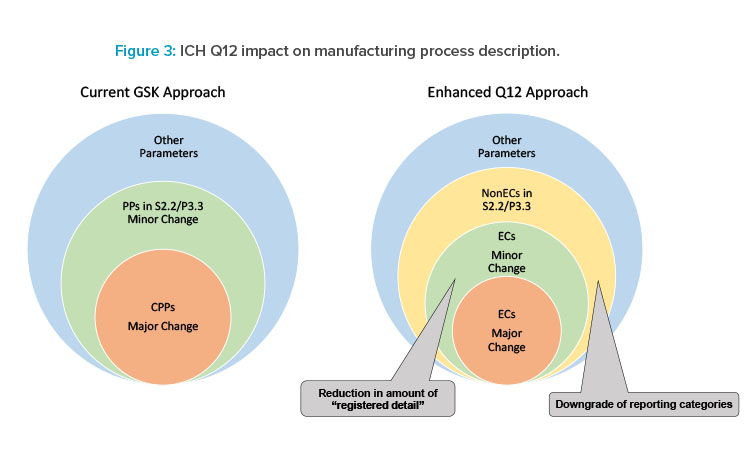
ICH Q12 provides an opportunity to reduce registered detail requiring regulatory action while providing regulators the necessary transparency to review a product’s process as illustrated in Figure 3. The enhanced Q12 approach will provide an opportunity to clearly define established conditions requiring regulatory action and potentially downgrading regulatory reporting categories for others. This leads to relevant information of established conditions and non-established conditions to be included in the Module 3 CTD sections S.2.2 and P.3.3 when the enhanced ICH Q12 approach is implemented. GSK continues to advance adoption of ICH Q12 tools to support applications in markets as Q12 implementation evolves globally.
Pfizer
Pfizer used an enhanced approach as described in ICH Q121 to propose established conditions and corresponding reporting categories in a PLCM document that included established conditions across the full scope of Module 3. The PAS submission was submitted as part of the US FDA Office of Policy for Pharmaceutical Quality’s established conditions Pilot Program.12 Pfizer’s science- and risk-based approach was based on rigorous risk assessments, overall control strategy, process understanding, product knowledge, enhanced analytical method development, and a robust PQS.14
The small molecule active ingredient synthesis included robust starting material controls and multiple in-process controls to ensure drug substance quality. A safety-based approach was used to determine reporting categories for specification established conditions to support the overall control strategy. The drug product used compendial excipients, direct compression, and a standard tableting process. The analytical methods to assess the drug substance and drug product included six different procedures for which established conditions were based on the method principle, method-specific performance criteria (i.e., validation criteria per ICH Q2,15 and higher-level method parameters.14
A comparison of the reporting categories in the PLCM versus current FDA guidance16 revealed several instances where the PLCM and the guidelines are aligned, as well as many established conditions that have reduced reporting categories. The latter are summarized in Table 2.
ICH Q8–Q11 provided the framework for science- and risk-based approaches; however, technical and regulatory gaps prevented the realization of postapproval flexibility. ICH Q12 provides the needed regulatory framework, and by leveraging the concepts therein,1 the approved PLCM increases flexibility and enables continual improvement of the product.14 Pfizer is currently exploring opportunities to implement ICH Q12 tools in global registration applications.
| Drug Substance Manufacturing Process |
Drug Product Manufacturing Process |
Analytical Performance | Specification |
|---|---|---|---|
| • Omission of recrystallization from the manufacturing process reduced from PA to NM. • Changes to 8 CPPs reduced from PA to NM. • Changes to 3 PPs that have very low risk to impact quality attributes reduced from NM to NL. • Changes to 24 noncritical process parameters in Steps 1 and 2 reduced from NM to NR. • Changes to 18 noncritical process parameters in the final step reduced from PA to NR. |
• Changes to the equipment using the same design and operating principle reduced from NL to NR. • Changes to tablet weight and film coating IPCs reduced from NM to NL. • Changes to screen aperture will be reduced from PA to NL. |
• Changes to method principle and performance criteria will be reported as PA. • Changes to 35 higher-level operational parameters (e.g., solvent system for HPLC) and the system suitability criteria will be reported as NM (no reduction). • Change to 6 slightly more detailed parameters (e.g., the wavelength of the analysis) reduced from NM to NL. • Changes to 65 parameters reduced from NM to NR. |
• Safety- and risk-based approaches were used to define reporting categories for raw materials critical IPCs and intermediate and drug substance specifications. |
Feedback on Implementation Progress
As individual companies have begun to implement established conditions and ICH Q12 concepts, it has become clear that there is some divergence in approaches and associated terminology that have been communicated to health authorities. Recent engagements between industry leads and health authorities have highlighted key issues faced by regulators and sponsors in implementing ICH Q12, and defined a potential path to a more harmonized approach to postapproval change management in the future.
One of the key challenges emphasized during the 2021 ISPE Challenges and Success of ICH Q12 Related Submissions webinar17 was associated with the importance of robust criticality assessments and effective communication of results and data to regulators in support of the proposed established conditions. Most of the companies surveyed have relied on the enhanced approach as defined in ICH Q12. Many companies have also found it necessary to update their risk assessment processes to be able to justify the appropriate reporting categories for those process parameters where a quality impact cannot be ruled out. Some companies have found it useful to introduce an additional intermediate criticality category of a KPP, while others employed “criticality continuum” approaches (e.g., based on a severity assessment) without introducing the KPP concept. Future alignment of terminology across industry will help improve consistency, as well as strike the appropriate balance in providing a sufficient level of detail in the dossier while maintaining life-cycle flexibility.18
FDA perspectives highlighted during the ISPE ICH Q12 webinar were in line with issues facing industry related to ICH Q12 implementation. Regulators emphasized the importance of effective and clear communication where “a shared understanding of the applicant’s intent, scope, and nomenclature is essential.18” The key takeaway from the FDA pilot experience was the varying approaches taken by applicants, with emphasis on the complexity of proposals put forth by sponsors often exceeding examples outlined in ICH Q12. Challenges related to existing products that were developed prior to ICH Q8, and therefore prior to formal criticality assessments for process parameters, provide an opportunity for ICH Q12 to address gaps associated with legacy products. To holistically address these challenges, FDA adoption and concurrent draft guidance for considerations in implementing ICH Q12 are currently in progress.
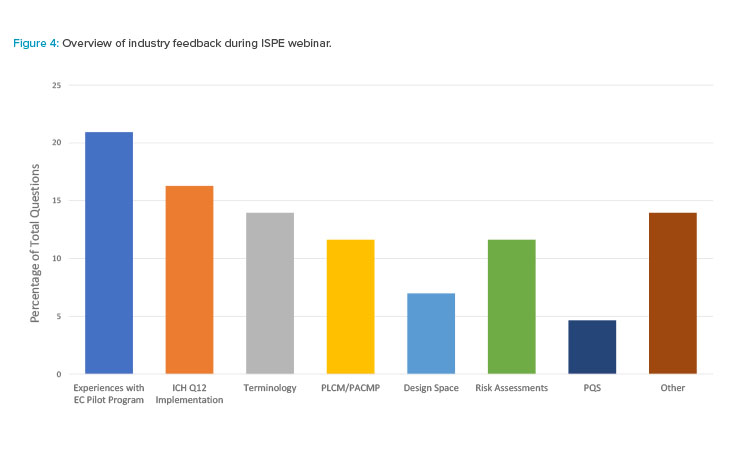
Industry participants had the opportunity to ask questions and provide live feedback to the presenters during the ISPE webinar. Seven key areas were identified as topics of interest:
- Design space
- Experiences with the established conditions pilot program
- ICH Q12 implementation
- PQS
- PLCM/PACMP
- Risk assessments
- Terminology
ISPE webinar participants were most interested in hearing about experiences with the FDA established conditions Pilot Program (Figure 4). FDA perspectives on common themes and what FDA found challenging, as well as the industry perspective on the overall pilot experience, were major themes. Implementation of ICH Q12, particularly from a regulator perspective, was the next most common topic. The status of implementation at the FDA, considerations regarding a harmonized approach across health authorities, and an understanding of how to implement ICH Q12 concepts for accelerated biologics program were of interest. Overall, these themes highlight areas of opportunity for future improvements as implementation of ICH Q12 progresses.
Conclusion
As described in the case studies, many companies have formed internal teams to implement the necessary adjustments to systems in preparation for formal implementation of ICH Q12. Initial experience with utilization of the regulatory tools has advanced collective knowledge that will help with future submission. These tools will enable and encourage increased transparency between industry and regulators.
However, challenges still exist, as flexibility in postapproval change management has not been fully realized. In certain cases, local guidance and regulation are not yet compatible with the ICH Q12 concepts, and thus, no implementation or partial implementation will result. Lack of global convergence and alignment on implementation approaches, as well as legislative constraints, will give rise to divergent implementation strategies among health authorities. There are existing submission pathway differences and diversity in timing of approvals. Lack of alignment for data expectations regarding necessary information and level of detail in the regulatory dossier is also a challenge and will need to be addressed. Ideally, the content of the PLCM document and PACMPs should be aligned and there should be global approval of one set of established conditions. Efforts to pursue global alignment of these ICH Q12 concepts will result in greater harmonization.
Worldwide adoption of ICH Q12 tools can provide a consistent approach to PLCM, with the potential for application in nonICH countries as well. As discussed, the harmonized approach to postapproval change management described in ICH Q12 will indeed “benefit patients, industry, and regulatory authorities by promoting innovation and continual improvement in the pharmaceutical sector, strengthening quality assurance and improving supply of medicinal products.”1
About the Authors





Acknowledgements
The authors would like to acknowledge the ISPE PQLI® ICH Q12 team for its contributions to the subject matter discussed in this article and the organization of the 2021 ISPE ICH Q12 webinar.


