Validation of Aseptic Processes Using Media Fill
Aseptic process simulation (APS) is essential for validation of an aseptic manufacturing process and is required by regulators to demonstrate the aseptic capability of such processes. A successful program of APS and aseptic manufacturing requires significant operator training, skills, and supervision; thorough maintenance; effective cleaning and disinfection; significant oversight of every aspect of the operation by quality assurance; and microbiological monitoring by quality control.
An overall validation of aseptic processing (as distinct from manufacturing process validation [PV]) is used to assess the contamination risk of an aseptic production process by simulating the manufacturing process using microbiological growth media instead of the drug solution. This is necessary in part because the sterility test used to release batches of sterile products has inherent limitations in detecting contaminated units in batches with low levels of microbial contamination, due to the limited number of samples that can be removed for destructive testing; this relationship has been evaluated statistically.1
Sterility assurance in aseptic processing requires contributing elements—such as the heating, ventilation, and air conditioning (HVAC) system, clean-room environment, material transfer, equipment, and manufacturing process steps, including sterilization processes and sterilizing filtration—to be qualified and validated as applicable and for personnel to be trained and qualified. Simulation of aseptic manufacturing processes using liquid microbiological growth medium (also referred to as media simulation or APS) is required by regulators to demonstrate the aseptic capability of these processes.
APS consists of three consecutive media simulations with designated personnel in the specific cleanroom environment, followed by repeat media simulations at six monthly intervals. Any media fill failures require thorough investigation and root cause analysis, and further media simulations may be required to complete the validation.
Aseptic processes are typically carried out in conventional cleanrooms with vial filling and stoppering in Grade A laminar airflow (LAF) in a Grade B background environment. The filling environment may be further protected within a restricted-access barrier system (RABS) with glove ports for access to the filling line. Alternatively, processing equipment for the critical steps may be enclosed in a glove box or isolator. Each of these systems enhances the filling environment’s sterility assurance but also presents challenges for material transfer, operator access, environmental monitoring, and APS.
Regulatory Expectations
Aseptic manufacturing and validation follow current GMPs and related GMP Annexes and Guidance. These pertain to the manufacture, validation (APS), and control of sterile products for injection (as well as eye drops and advanced therapy medicinal products). Current guidelines come from the European Union/Pharmaceutical Inspection Convention (EU/PICS), China (2010) GMP (NMPA), United States Food & Drug Administration (US FDA), and World Health Organization (WHO). 2, 3, 4, 5, 6, 7, 8, 9, 10, 11,12, 13 They may reference related International Organization for Standardization (ISO) and Parenteral Drug Association (PDA) standards,14, 15, 16, 17 such as those relating to cleanroom air-cleanliness classification and particle monitoring.17
Because of the high safety risk profile for parenteral drug products, the protocols, results, and reports for APS form an integral part of regulatory submissions for such products, meaning they are included in investigational new drug (IND) applications, new drug applications (NDAs), and marketing authorizations (MAs). Ancillary documents such as training records, environmental monitoring reports, deviations, and investigations are key topics of scrutiny during facility inspections, as well as the qualification of facility, the equipment and utilities, and the process validation.
The expectation in APS is twofold. First, it must achieve three consecutive media batches that meet target acceptance criteria. Second, the solution filtration process must be validated against a microbial challenge with 107 colony-forming units per square centimeter of filter medium (using Brevundimonas diminuta, a small-celled Gram-negative bacterium suspended in the drug solution).
Examples of media fill run sizes and acceptance criteria for APS that have been incorporated in GMP Annex6 and Guidance10, 11 include:
- When filling less than 5,000 units, zero contaminated units should be detected. A contaminated unit is considered cause for revalidation following an investigation.
- When filling 5,000 to 10,000 units, one contaminated unit should lead to an investigation, including consideration of a repeat media fill. Following investigation, two or more contaminated units are cause for revalidation.
- When filling more than 10,000 units, one contaminated unit should lead to an investigation, and two or more contaminated units are cause for revalidation.
APS Considerations
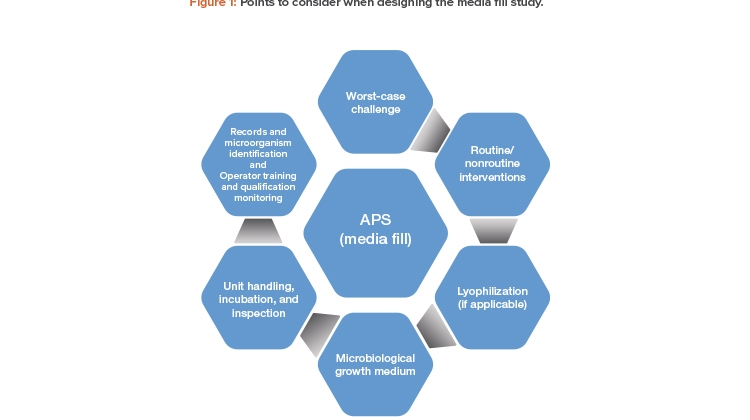
The following is an overview of points to consider when designing the media fill study for an aseptic manufacturing process.
Worst-Case Challenge
APS should mimic, as closely as possible, all aspects of the aseptic manufacturing process and should involve a “worst-case” approach as a challenge to the robustness of the aseptic operations. The “worst-case” should be defined with supporting rationale.
Risk assessment principles should be used to determine the worst-case challenges related to line speed, container size, batch size, hold time, con-figurations, and operating conditions.
Some examples of worst-case challenges :
- Filling process
- Aseptic assembly of equipment and aseptic connections prior to commencement of filling
- Slowest filling speed with widest opening vials/containers
- Maximum filling volume for small vials/containers, due to handling difficulty that can result in more interventions
- Maximum batch filling duration (may include lyophilizer loading and door opening duration)
- Operator fatigue as contamination risk
- Operating conditions
- Maximum number of personnel in aseptic area
- Shift changes, personnel changes, and operator breaks
- Hold time
- Equipment/room clean hold time
- Equipment sterilization hold time
Routine and Nonroutine Interventions
Interventions to be included for simulation in the media fill protocol include routine and nonroutine manipulations by operators. The regulatory expectation is that interventions included in APS should be compliant with current GMPs, and APS must not be used to justify poor aseptic practice or equipment design.
Routine interventions include charging stopper and seal hoppers, removing jammed stoppers or toppled vials, taking environmental monitoring samples (settle plates, active air samples, and contact plates), and checking in-process control samples (e.g., manual weight checks). Routine interventions should be performed as described in the production standard operating procedure (SOP) or the batch record or environmental monitoring SOP. Procedures to be followed in the event of machine jams and spills may include partial line clearances, including removal of exposed units.
Nonroutine interventions may include changing the filling nozzles or handling unexpected events, such as breakdown maintenance, line stoppages, machine adjustments, and material transfers. Interventions can also be grouped by access point, and their risk assessed so that worst-case (highest risk) interventions are included in the study.
Lyophilization
EudraLex Annex 1 (2009)6 states, “The process simulation test should imitate as closely as possible the routine aseptic manufacturing process....” It is unlikely that the exact lyophilization cycle for the product can be replicated during media simulations due to the constraint of maintaining the media to support microbial growth. Deviation from the production cycle must be justified. For example, if the recommended temperature range for media is 5°C to 25°C, the chamber pressure, normally 100 to 200 mbar, should not be lower than the equilibrium vapor pressure of the media at the loading temperature to avoid boiling away the media and to avoid overconcentration of media, which could adversely affect the recovery and growth of microorganisms.
The chamber dwell time during APS does not impact risk because the higher chamber pressure required to avoid boiling of media does not require the use of a pressure control (gas injection) system. In the absence of airflow transport mechanism and turbulence, the chamber dwell time becomes immaterial during APS. Based on risk analysis, the aeration or vacuum-break step in the lyophilization cycle may have higher risk of contamination because it involves air turbulence18 and the possibility of entrained particles entering the containers. Because the application of full vacuum is not possible during APS, multiple partial vacuum steps should be considered to simulate the worst-case aeration. The media volume in the vials before lyophilization must ensure the wetted surface of the container mimics the production case.
Media simulation of the lyophilization step could involve loading the required number of media-filled vials as per the routine commercial production procedures, while assuring the time that the door is open to the cleanroom environment is at least as long as the maximum time incurred when loading a commercial batch of product.
Once the modified media lyophilization cycle has been completed, the chamber vacuum should be broken using sterile-filtered compressed air so that all units are stoppered under pressure to avoid inhibiting microbial recovery and growth. (Sterile-filtered nitrogen gas should not be used to break the vacuum unless a specific anaerobic media simulation is undertaken.)
Microbiological Growth Medium
Media for microbiological recovery and growth are defined in pharmacopoeia—such as the United States (USP), European (Ph. Eur.), Chinese (ChP), and Japanese (JP) Pharmacopoeia—and should be made and sterilized according to the manufacturer’s instructions. The media used in APS for filling sterile, depyrogenated containers is generally tryptone soya broth (TSB), or soybean casein digest medium (SCM), which supports recovery and growth of viable aerobic microorganisms. Anaerobic growth medium such as fluid thioglycolate medium (FTM), which supports recovery and growth of obligate or facultative anaerobic bacteria, may be considered under special circumstances (e.g., where the product solution is to be filled into nitrogen-flushed vials).
The growth medium, supplied as a dry powder, is a critical material for APS. It is recommended that the manufacturer is qualified and monitored as an approved supplier; a growth promotion certificate may be obtained with every batch. Prior to release for use, batches of the media to be used for APS should be reconstituted and sterilized; then samples should be subjected to quality control testing for growth promotion by inoculating with ≤ 100 colony-forming units of representative compendial strains of microorganisms. Microorganism strains from environmental monitoring may be included in the growth promotion test.
Unit Handling, Incubation, and Inspection
After filling, stoppering, and sealing, 100% visual inspection is performed for defects such as the presence of visible foreign matter, high or low fill volumes, and damaged vials, stoppers, or seals. Such defective units would be normally removed (rejected) from product batches, but in the case of APS batches, such defective integral units must be retained and all such containers must be incubated. If filled containers are broken or otherwise damaged so that they are nonintegral and potentially contaminated, they must be recorded and reconciled with the batch record quantities. All appropriate media fill container units must be incubated.
The incubation conditions selected are optimal for recovery and to allow for detection of both slow-growing and normal contaminating organisms, i.e., adequate to detect microorganisms that might otherwise be difficult to culture. The incubation conditions used generally are 20°C to 25°C for seven days (lower temperature first) followed by 30°C to 35°C for a further seven days.
Containers are typically incubated on their sides, and while subjected to each incubation temperature, turned at least once to ensure that the entire interior surfaces of the vials and the stoppers are contacted by the growth medium.
Records (chart printouts or electronic records) of the incubation conditions must be maintained, including the date and time of incubation commencement, turning of vials, transfer to the second incubator, and further turning and completion of incubation. Incubated vials must be inspected by operators qualified to distinguish sterile vials (“no growth”) from vials showing microbial growth (surface pellicle or turbidity in the solution). A small number of sterile (“no growth”) vials should be selected from the incubated vials for use as after-test growth controls; these vials are then inoculated with ≤ 100 colony-forming units of the compendial microorganism strains mentioned previously, and incubated, followed by inspection for positive microbial growth.
Environmental Monitoring
During APS, all routine and normal processes (such as cleaning, disinfection, and maintenance) should be continued to maintain the cleanroom environment in qualified status. This includes particulate and microbiological environmental monitoring, which can demonstrate that the specified clean-room environment conditions are maintained. These monitoring results may provide key information for the investigation of a failed media run.
Particulate monitoring during aseptic product filling and APS consists of continuous monitoring for particulates in the < 0.5 μm and < 5.0 μm ranges, using a particle sampler attached to an isokinetic probe located near to the point of fill in the Grade A area. A permanent record of the particle counter's printout (or certified true copy if the printout is on thermal paper) must be attached to the batch record for the product fill or APS batch. The regulatory/action limits for the monitoring, per m3 air volume, are not more than 3,520 particles in the < 0.5 μm particle size range and not more than 20 particles in the < 5.0 μm range.
The microbiological methods used should be described in an SOP, including a map of the locations at which the samples are to be taken or plates exposed. Each batch of environmental sampling plates must be tested for sterility and growth promotion capability against the recommended compendial strains of microorganisms before release for use.
The methods used for environmental monitoring are stated in China GMP3 and EudraLex, current Annex 1:6 active air sampling (1 m3 sample volume) onto 90 mm agar plates; settling plates 90 mm in diameter, with exposure up to 4 hours (if the APS or production filling lasts longer, new settling plates must be exposed for each subsequent 4-hour period); surface contact plates 55 mm in diameter (in which the plates are contacted against machine surfaces or cleanroom walls, floors, or operator gowns); gloved-finger samples performed by cleanroom operators during the filling period and upon leaving the cleanroom, taken by contacting four fingers and thumb onto the surface of a 90 mm tryptone soya agar (TSA) settle plate.
Media is usually TSA for viable aerobes or sabaroud dextrose agar (SDA) for fungi (molds) and yeasts. Surface contact plates may be TSA, usually incorporating a neutralizing agent to counter detergent residues from the sampled surfaces. Agar residues are removed from the sampling locations by wiping with 70% alcohol.
The expected (regulatory) action limits for the microbiological monitoring results of the Grade A cleanroom areas (Grade A LAF in Grade B back-ground; RABS; isolator), including during APS, in colony-forming units are tabulated in China GMP3 and EudraLex, current Annex 1.6 Adjacent Grade B, C, or D cleanrooms through which operator gowning and material transfer for the APS occur should also be monitored; the stated regulatory (action) limits for these cleanroom grades are also included in the China GMP3 and EudraLex, current Annex 1.6 The frequency of monitoring Grade C and D cleanrooms is to be determined based on quality risk assessment because such monitoring at the time of an APS may help investigate any discrepancy or failure.
Records and Microorganism Identification
In APS batches, the numbers of colony-forming units recorded on the environmental monitoring plates in Grade A (LAF, RABS, or isolator) and Grade B areas should be recorded. An isolate should be taken from each visually distinct microbial colony and identified by species using available biochemical and/or nucleic acid identification methods so it can be compared with organisms in contaminated units that arise during the APS. This information will be critical in investigating and determining corrective actions in the event of an APS media fill that exceeds acceptance criteria. Environmental samples (those with colonies) from Grade C and D cleanrooms should be enumerated and preferably also identified, as the information regarding the numbers, species, and locations of contaminating microorganisms may prove crucial in the investigation and resolution of a failed media fill.
APS consists of three consecutive media simulations with designated personnel in the specific cleanroom environment, followed by repeat media simulations at six monthly intervals.
Operator Training and Qualification
Prior to APS batch manufacture, operators performing APS must be trained in relevant procedures, including cleanroom gowning, aseptic connections, and correct cleanroom behavior, as well as in product-specific manufacturing procedures. All staff qualified to work in the area, including maintenance personnel, need to be included in APS.
Relevant training points:
- Sterile materials and equipment should be handled only with sterile instruments, such as forceps. Between uses, instruments should be protected from contamination.
- After initial gowning, sterile gloves should be regularly sanitized by spraying with a qualified sanitizing agent such as sterile 70% isopropyl alcohol (IPA) to minimize the risk of contamination. Personnel should not directly contact sterile products, containers, components, or critical surfaces.
- Rapid movements create turbulence in the critical environment, disturbing LAF and the integrity of sterile environments, and entraining particles. Operator movements should be slow and deliberate.
- Aseptic operators should not disrupt LAF designed to protect critical surfaces. When performing aseptic manipulations (such as making aseptic connections, removing samples, or retrieving fallen or jammed components from a filling line), operators should be trained to approach the location slowly and deliberately from the side whenever possible.
- After initial theoretical training, aseptic training operators should be allowed to practice their movements in a mock-up or nonsterile practice environment before being permitted to participate in operations in the cleanroom environment.
Media Fill Failures and Root Cause Determination
A key step in the investigation is identifying microorganism(s) species in positive media vials and any colonies appearing on environmental monitoring plates, particularly those from the Grade A/B environments, including from RABS/isolator monitoring. Identification of species from colonies on plates exposed in the lower-grade adjacent cleanrooms, through which materials or personnel have accessed the filling rooms, may also be crucial.
The review of the deviation should encompass the preparation and manufacturing processes—including cleanroom cleaning and disinfection, components and materials sanitization/sterilization and transfer processes, HVAC and cleanroom operating parameters during the filling period, filtration process and integrity tests, filling operation, stoppering and capping equipment, and taking and transferring in-process or environmental samples. The review should focus on documentation, including any deviations or atypical events, but may also include a review of CCTV records of the filling rooms and operations and documented interviews with operators. Review should also include recent engineering work or prior media fill batches.
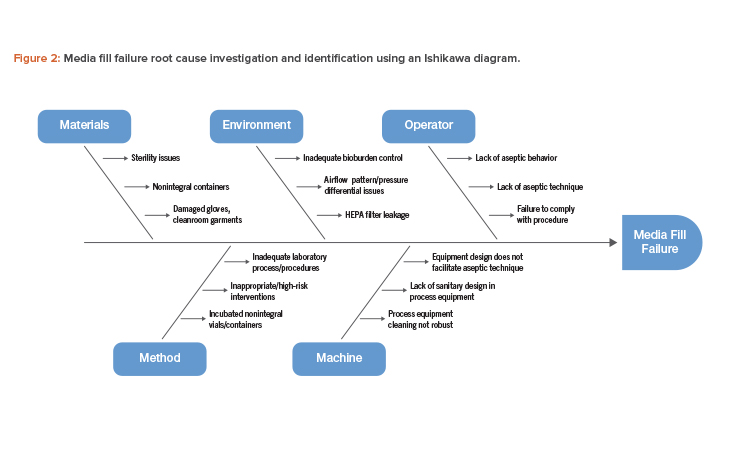
An Ishikawa diagram showing cause-and-effect links to a specific failure is a useful tool that can be used to investigate and identify the root cause of a media fill failure (see Figure 2).
Based on the potential root cause interactions identified in Figure 2, investigating the possible failure modes and corresponding risk mitigation measures will be necessary (Table 1).
In the investigation, different possibilities may provide the evidence to support root cause determination, such as the ability to match the identification of an environmental isolate from the current (or recent) batch with the identity of the contaminating organism in the failed media units, or a significant processing discrepancy or error or equipment failure.
Case Study
In a sterile injectables manufacturing plant, a routine media fill showed growth in one vial. The microorganism was a micrococcus, typically associated with human skin, attributed to an engineering intervention using an unsterilized tool and not reflective of normal practice. A repeat media fill was done, which also showed growth in one vial with no obvious root cause. Manufacturing of product was put on hold. Following an investigation, it was noted that the APS included approximately 80 interventions to simulate any possible activities that might be required in normal production. However, in normal production, far fewer (< 20) interventions occur routinely. Therefore, it was concluded that the process may have been excessively stressed and was not representative of the commercial process being simulated. Three further media fills were initiated, of which the first media fill showed growth in one vial.
The investigation using RNA ribotyping identified that the microorganism in all three media fills showing growth was the same—a micrococcus. Microbial testing showed that one operator tended to shed greater numbers of skin particles than other operators, including this microorganism. The investigation also identified variability in how materials were passed into the sterile core, potentially providing a route of ingress.
A risk assessment was carried out to determine any safety issues arising from the sporadic low-level contamination in the process. It was concluded that based on the nature of the microorganism, the sterility assurance levels achieved by the process, and the regulatory guidelines, the safety risk was low. However, it was now obvious that the process was not operating in a validated state. No further batches of the product were manufactured until the process was shown to be in a validated state, as evidenced by three successful media fills. Members of a sterility assurance expert group from the wider company assisted during the investigation. The plant ensured that the necessary remediations identified during the investigation—reallocation to other duties of the “shedding” operator and reduction in number of interventions simulated per media fill (the interventions were divided into three groups, one group to be included in each of three media simulations)—and the potential contributory aseptic practices were revised and operators re-trained before conducting three successful media simulations to revalidate the process.
| Description | Possible Failure Mode | Risk Mitigation | |
|---|---|---|---|
| Operator | Aseptic behavior |
|
|
| Aseptic technique |
|
||
| Compliance to procedure | Tasks not performed according to procedure | ||
| Machine | Equipment design | Equipment design not facilitating aseptic interventions, and increased risk of contamination during intervention |
Design qualification; modifications may be required to mitigate the risk |
| Sanitary design | Inadequate sanitary design in process equipment leading to cleanability issues |
||
| Process equipment cleaning | Process equipment cleaning procedure not robust | Design a robust cleaning procedure considering the type of soil, cleaning parameters, and the use of appropriate cleaning detergent, if required |
|
| Reduced cleanability of stainless-steel surfaces due to corrosion, which could lead to formation of biofilm |
Periodic maintenance of stainless-steel surfaces to minimize risk of corrosion | ||
| Environment | Bioburden control | Inadequate cleaning and disinfection program for cleanroom surfaces |
Design a robust cleaning and disinfection program using sanitizers, disinfectants, and sporicides |
| Airflow pattern/pressure differential |
|
|
|
| Leakage | Leakage in HEPA filter | Periodic maintenance and leak tests | |
| Method | Laboratory process/procedures | Laboratory process/procedures inadequate | Review and remediate laboratory procedures to minimize errors |
| Inappropriate/high-risk interventions |
High-risk interventions disrupt unidirectional (laminar) airflow, and thus increase the risk of contamination |
Smoke studies to be conducted and evaluated for risk of contamination (turbulence) for each intervention |
|
| incubated nonintegral vials/ containers |
Failure to spot nonintegral vials/containers | Training of inspectors on nonintegral vials/containers | |
| Material | Sterility issues | Parts/packaging components not sterile due to sterilization process issues |
Verify sterilization processes meet acceptance criteria and assess impact of any excursions |
| Nonintegral containers | Sterility of parts/packaging components compromised after sterilization, prior to usage |
Ensure sterile parts/packaging components are protected from contamination through the use of appropriate protective barrier |
|
| Damaged gloves, cleanroom garments |
Nonintegral vials/containers not segregated | Training on inspection of nonintegral vials/containers prior to incubation | |
| Damaged gowns and gloves can increase risk of contamination | Confi rm the integrity of the gowns and gloves visually |
| NO. | Area | Observations |
|---|---|---|
| 1 | APS design |
|
| 2 | Operational | Failure to:
|
| 3 | Root cause analysis | Failure to:
|
| 4 | Personnel | Poor aseptic technique and practices:
|
Potential GMP Discrepancies During Media Simulations
Given the enhanced frequency of regulatory inspections in companies where aseptic manufacturing is used and the growth of monoclonal antibody and other biological products requiring aseptic filling, there are many examples of GMP failures and APS issues. Some typical examples that have appeared in warning letters and summaries by regulators are provided in Table 2.
Conclusion
APS with microbial growth media is an integral part of an aseptic manufacturing operation. The design of the APS must take into consideration various operating parameters to avert a worst-case scenario for the media fill challenge. Such parameters can be determined by risk assessment, and typically include the container-closure configuration, batch size, operating conditions, and interventions. The risks involved with individual interventions need to be identified, assessed, and mitigated to minimize contamination risk. Equally important is a team of highly trained and competent operators that have knowledge of microbiology and aseptic technique and practices; a sound and effective cleaning and disinfection program for cleanrooms; regular equipment cleaning and maintenance; and cleaning and sterilization processes. Attention to such considerations ensures a robust and successful APS program.
About the Authors




