Is a Globally Harmonized Quality Overall Summary Possible?

The International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guideline on Registration of Pharmaceuticals for Human Use (M4)1 offers advantages in the consistent format of the registration dossier using the Common Technical Document (CTD). However, it does not deliver a comprehensive view of the overall manufacturing control strategy or a means of understanding and managing the quality of the product throughout its life cycle. As a result, several regulatory authorities that have implemented the CTD format have also insisted on supplementary quality summary documentation that exceeds ICH requirements, and, in effect, creates divergent expectations for chemistry, manufacturing, and controls (CMC) content. A single global quality overall summary (QOS) format could clearly convey a holistic view of a product’s control strategy and improve the efficiency and economy of the regulatory review of an application while providing a way for the applicant and reviewer to align on a product life-cycle management plan.
The current structure and format of the QOS does not provide a mechanism to holistically integrate the elements of the control strategy in the CTD. This, and the need for review efficiency, has prompted several regulatory authorities to implement their own unique summary document formats and requirements to facilitate their reviews. These redundant summary documents diverge in scope and demand varied information be included in the registration application for different countries, which can result in a delays in submission, approval, and availability of critical medicines to patients worldwide.
This article endeavors to capture the rationale and purpose for the multitude of CMC summary documents required in various countries and offers a proposal for how a more holistic and comprehensive QOS could be structured while leveraging the concepts and tools outlined in ICH Q12, specifically the sections on “Established Conditions” (ECs) and the “Product Lifecycle Management” (PLCM) document.
A consistent content and format for the QOS could lead to harmonized marketing authorization (MA) filings globally and streamlined regulatory authority assessments of quality information. The proposal for a more holistic and comprehensive QOS will provide a mechanism for industry to convey a holistic view of their ECs and control strategies and could increase consistency and efficiency in regulatory decision-making and actions, which would facilitate timely approvals and accelerate access of new drugs to patients.
Regulatory Landscape
ICH has now grown to include 17 members, and guidelines are being adopted by a growing number of regulatory authorities.2
The ICH website provides a summary of the self-declaration of the regulator regarding the conclusion of the implementation process. A status of “implemented” implies the process of implementation is completed. The following agencies3 have declared implementation of the ICH Guideline: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Quality ICH M4Q (R1)1 and M4Q Q&As (R1)—Questions & Answers CTD on Quality:4
- Brazil National Health Surveillance Agency (ANVISA), Brazil
- EC, Europe
- Food and Drug Administration (FDA), United States
- Health Canada, Canada
- Health Sciences Authority (HSA), Singapore
- Ministry of Food and Drug Safety (MFDS), Republic of Korea
- Ministry of Health, Labour and Welfare/Pharmaceutical and Medical Device Agency (MHLW/PMDA), Japan
- National Medical Products Administration (NMPA), China
- Swissmedic, Switzerland
- Taiwan’s Food and Drug Administration (TFDA), Chinese Taipei
The Quality sections of the CTD include detailed CMC information in Module 3 (M3) and a summary of the CMC information in Module 2 (M2, or QOS).
![Figure 1: The CTD triangle (© ICH) [6].](/sites/default/files/2022-08/0922_PE_SO_CoverStory_02.jpg)
While the current CTD structure offers some advantages in the harmonized format of registration dossiers, challenges with the format and content of the QOS exist in several countries as reflected by the requirement for specific templates and/or additional content not required by ICH M4Q.
Challenges With the QOS Structure
The ICH M4 (R4) guideline, Organization of the Common Technical Document for the Registration of Pharmaceuticals for Human Use,5 describes the CTD as five modules. As shown in Figure 1, the CTD is the format implemented in multiple ICH regions. ICH M4Q (R1) provides guidance on how to structure both M3 and the QOS, which links to more detailed quality data and information in M3.
Options for granularity are provided in the ICH M4 (R4) guideline for the QOS. These options range from a single QOS containing a summary of all quality information to a QOS that is systematically organized in alignment with the granularity of M3. There is one M2 section corresponding to each major M3 section and appearing in the same sequential order, as illustrated in Figure 2.
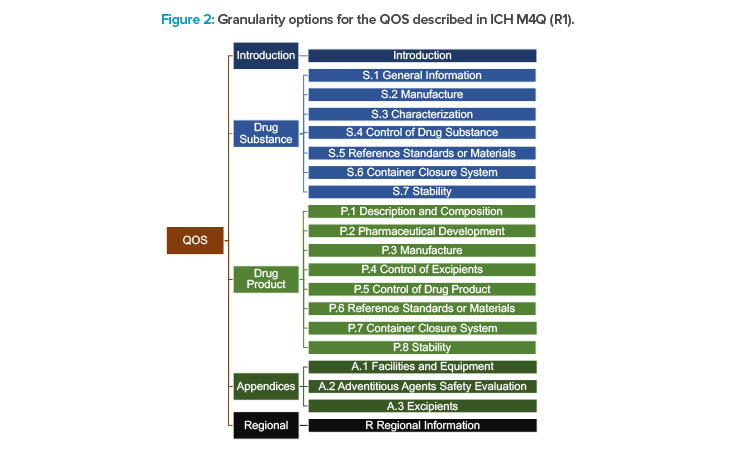
Regardless of how the quality information is summarized, there should be no CMC content included in the QOS that is not also included in the M3 Quality CTD sections. It is important to note that the QOS content is generally not maintained after the initial marketing authorization approval or throughout the life cycle of the product. The QOS document(s) that accompany the CTD application is/are typically copied and pasted from the data in the corresponding M3 sections and may be used by some regulators as the basis for their initial review template. From an industry perspective, the QOS has not been considered very useful to present a holistic view of a product’s control strategy.
Ongoing QOS-Related Initiatives
Several ongoing initiatives should be considered in the development of a harmonized QOS.
US FDA White Paper
In 2018, the FDA authored a white paper indicating the agency’s desire to use the QOS as a tool to “improve the efficiency and quality of the regulatory assessment” and to “communicate essential aspects of the application.”7 As the typical QOS is copied and pasted from M3 sections, it presents a very segmented and incomplete view of a product’s control strategy. Ideally, the alignment of the QOS content across the ICH regions would focus the application assessment on the importance of the drug substance and drug product control strategies and summarizing the regulatory commitments.
ICH M4Q Revision
In 2020, the ICH Assembly supported a proposal to revise ICH M4Q: The Common Technical Document for the Registration of Pharmaceuticals for Human Use: Quality–M4Q (R1), Quality Overall Summary of M2, M3: Quality.8 The M4Q (R2) Informal Working recently approved its Concept Paper and Business Plan.9, 10 Specifically, the working group will reorganize the CTD Quality sections in M2 and M3 to capture submitted information on drug substance and drug product specifications, characterization and control of impurities, analytical testing and validation, stability data, manufacturing process and unit operations, associated manufacturing facilities, and container closure system in an organized structure consistent with modern pharmaceutical manufacturing and regulatory guidance.
Structured Product Quality Submissions (SPQS)
Algori, et al., suggested structured content management (SCM) as an opportunity for enhancing review and organization of documents by providing the framework for authoring content that is easy to adapt into multiple documents and standardize.11 Using XML-based systems for the management of CMC data provided in the CTD in M2 and M3 quality sections have also been proposed by some regulators to facilitate a more highly structured assessment as compared to today’s narrative approach to submission and review. In June 2018, the FDA announced a knowledge-aided assessment and structured application (KASA) initiative as a new review platform to modernize generic drug review from a text-based to a data-based assessment and to promote robust generic entry. 12
Leveraging structured content management could potentially streamline the summary of quality data in regulatory submissions and was also supported as a new topic proposal at the ICH Assembly virtual meeting in 2020.8 Structured data using XML-based systems have been used to enhance regulatory review and to harmonize the organization of registered content. This allows for information classification and re-use of content that contains descriptive metadata to aid in structuring and presentation of data elements. A structured platform could also enable automated analysis of some portions of the application, which would save time and ensure consistency.
A successful implementation of any structured product quality submission for CMC content will require significant global collaboration and alignment, as well as coordination with other ongoing proposals to revise CTD M2 and M3. When proposing to harmonize the global regulatory requirements for CMC summary documents, the ongoing efforts to implement SPQS and revise the ICH M4Q M2 and M3 content guideline(s) must both be considered. Ideally, the use of XML for the management of CMC data provided in M3 could facilitate the generation of any format for a summary document required by a regulator as a review template.
ISPE Regulatory Quality Harmonization Committee (RQHC)
The mission of the ISPE RQHC North America group is to anticipate, engage with, and facilitate technical implementation of regulatory guidance and expectations in North America to the benefit of ISPE members and stakeholders.13 A subcommittee of this group is developing proposals on the content and structure of a risk-based QOS that could ultimately standardize CMC terminologies and submission standards for control strategy harmonization and cloud assessment.
Implementation of ICH Q12
The ICH Q12 guideline, Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management, is in the process of implementation for ICH members.14 Once implemented, the framework described in Q12 “to facilitate the management of post-approval CMC changes in a more predictable and efficient manner”14 and the tools and approaches (including ECs and the PLCM document) can play an important role in the development of a comprehensive QOS. ECs, which are defined as “legally binding information considered necessary to assure product quality” allow a clear understanding between the marketing authorization holder and the regulatory authorities on which elements of the control strategy assure product quality, and which information can be designated as supportive.14
The PLCM document summarizes the ECs, and once approved, serves as an agreement between the market authorization holder and the regulatory authority on the reporting category for future changes to those ECs. In instances where ECs are being proposed across the whole of M3, the PLCM document, along with a narrative that justifies the overall control strategy, could be used to form the basis of a comprehensive QOS that provides an accurate record of key data necessary to assure product quality and documents the reporting categories necessary to change it. The option to include the ECs within the context of the PLCM document in a single global format could also eliminate the need for unique regional CMC summary documents.
Evaluation of Regional CMC Summary Document
Several unique summary documents are required per some regulatory authority regulations that (a) may not follow the standard ICH QOS CTD format; (b) are above and beyond the QOS CTD requirements; (c) represent commitments or ECs; and (d) must be maintained throughout a product’s life cycle. Some of the more commonly known types of compliance summary documents include the Canadian Certified Product Information Document (CPID), the Japan Application Form (AF), and the Russian Normative Document (ND). Depending on the country, the level of M2/3 content required in the respective summary documents may differ.
CMC Requirements in Bespoke Compliance Summary Documents
CMC requirements for compliance summary documents were compared for this article for several countries, noting that some regions leveraged the same templates/requirements across several countries. For each country in scope of this evaluation, the following questions were addressed: (a) why do the regulators require CMC summary information in their specific format?; (b) is the submission of CMC information in this format considered mandatory?; (c) is the summary document considered a compliance document/must it be maintained after approval?; (d) what quality information is required in the compliance summary document?; and (e) is the quality information required in the compliance summary document beyond the ICH M4Q M3 information required?
A summary of the results from this evaluation is provided in Table 1 and Table 2. The summary in Table 1 is based on an interpretation of the respective country guidance documents for the summary documents, where available. A summary of the CTD M3 sections that would contain the technical content required in each of the regional compliance summary documents is provided in Table 2. In contrast to the segmented ICH M4Q(R1) QOS in Figure 2, the proposed QOS would provide the holistic view of the entire dossier and would highlight risks and provide links to more detailed information in M3. It would bring together all of the elements of the development experience and culminate in a comprehensive picture of the overall control strategy and cohesive narrative on how the ECs were determined and justified.
The majority of the CMC compliance summary documents require information from CTD sections that contain information as described in Appendix 1, Table A of ICH Q12,14 e.g., nomenclature, structure, manufacturer, manufacturing process, which is reflected in the “Proposed” column in Table 2. Here is a brief summary of the purpose and required information (including information not described in ICH Q12 Appendix 1, Table A) for each CMC compliance summary document.
- The Canadian CPID is a condensed summary of the key quality (drug substance and drug product) information provided in NDS applications. In addition to the compliance information, the CPID also includes drug substance general properties, impurities, process validation, and stability information, but it does not include drug substance nomenclature, control of materials, control of critical steps and intermediates, analytical procedures, reference standards, or drug product excipient information, analytical procedures, or reference standards content.
- The China MPIS is drafted by the applicant, reviewed with the initial marketing application, and then issued by the NMPA with product approval. It includes key compliance information on the drug substance (specifications and methods) and drug product (formulation, manufacturing process, specifications, methods, and container/closure).
- Although no specific regulation or guidance could be found on the requirements for the Republic of Korea CMC Summary, the authors’ experience through the receipt of regulatory queries has indicated this document is required. The Republic of Korea’s summary document primarily contains drug substance general information and specifications, drug product composition, excipient information, manufacturing process, container closure, and stability conclusions. The Republic of Korea also has a unique requirement to include the storage condition for non-compendial excipients in their compliance summary document.
- The South Africa SCoRE document is a relatively new requirement from the South Africa Health Products Regulatory Authority and is intended to facilitate review of new applications and to reduce evaluation time. In addition to the compliance information, the South Africa SCoRE document also includes drug substance general properties, elucidation of structure, impurities, and batch analysis content and drug product pharmaceutical development, process validation, batch analysis, and stability content but does not include drug substance, control of critical steps and analytical procedures or drug product excipient, or analytical procedures content.
- The Japan Application Form (AF) is a summary of many of the M3 sections, especially those focused on manufacturing commitments. Importantly, the Japan AF includes information for each manufacturing parameter to facilitate future post-approval changes; for each parameter’s target or set value, the use of or around each value indicates whether a future change to this parameter would be a minor change notification or partial change application, respectively. In addition to the AF, the PMDA also requires specific tables (Module 1.13, Table 1 and Table 2), which describe and justify the designation of manufacturing process parameters and acceptable ranges, which also facilitate postapproval changes. The CMC content in the Japan AF is the most closely aligned with the compliance information listed in ICH Q12 and with the content of the proposed QOS.
- The EAEU Quality Normative Document (QND) is different from the summary documents being discussed and includes not only a list of quality characteristics of the product but also quality control methods for a medicine, intended to facilitate local testing. Given the QND’s specific purpose, the proposed QOS may not readily replace this particular compliance summary document.
- The WHO QOS product dossier template is required by multiple Africa/Middle East (AfME) markets, as well as by Pakistan. The WHO QOS is a comprehensive document, with content the same as the required by ICH M4Q(R2), but in a different format and in a different template. Therefore, the WHO QOS could easily be replaced by the proposed QOS.
In general, the compliance summary documents summarized in Tables 1 and 2 are required in addition to what is required in ICH M4Q (R1) as part of the initial marketing application. These compliance summary documents are primarily intended by the various regulatory authorities to facilitate their review of the initial marketing application, and also, importantly, to facilitate the review of subsequent postapproval changes by documenting key quality information (e.g., commitments or ECs) in a standardized and summarized format. As noted in Table 2, except for the WHO QOS document which contains CMC content exactly in accordance with ICH M4Q (R1), the CMC content in the remaining compliance summary documents are a subset of the content required by ICH M4Q (R1).
| Country/Health Authority |
Name of CMC Compliance Summary Document |
Rationale for Unique Summary Document Format | Mandatory? | Must Be Maintained After Approval? |
|---|---|---|---|---|
| Canada/Health Canada | CPID: Certified Product Information Document – Chemical Entities (CPID-CE)15 |
|
Yes | Yes |
| China/NMPA | Drug Product Manufacturing Process Information Sheet (MPIS)/ Annex 2716 |
|
Yes | Yes |
| Japan/PMDA | Application Form, AF17 |
|
Yes | Yes |
| Republic of Korea/MFDS, formerly known as the Korea Food and Drug Administration (KFDA) |
CMC Summary Document8 18 |
|
Yes | Yes |
| South Africa/South African Health Products Regulatory Authority (SAHPRA) |
Summary of Critical Regulatory Elements (SCoRE)19 |
|
Yes | Yes |
| Eurasian Economic Union (EAEU)1 /Ukraine Uzbekistan |
Quality Normative Document20 |
|
Yes | Yes |
| Ethiopia, Gulf Cooperation Council (GCC)2, Ghana, Nigeria, Pakistan, Rwanda, SADC3/Various |
World Health Organization (WHO) QOS5 21 |
The QIS and QOS:
|
Yes but some markets may accept ICH M4Q M2 format |
Varies6 |
| Ghana, Nigeria, and South African Development Community (SADC)3 /Various |
WHO quality information summary (QIS)522 |
|||
| ICH Countries/Various | ICH M4Q Module 2 ICH M4Q (R1)1 Core ICH Q12 Guideline ECs, PLCM,4 and annexes 14 |
|
No | Yes, if filed |
- EAEU includes the following markets: Armenia, Belarus, Kazakhstan, Kyrgyzstan, and Russia.
- GCC includes the following markets: : Bahrain, Kuwait, Oman, Qatar, Saudi Arabia, and the United Arab Emirates.
- SADC includes the following markets: Angola, Botswana, Democratic Republic of Congo, Eswatini, Lesotho, Madagascar, Malawi, Mauritius, Mozambique, Namibia, Seychelles, South Africa, Tanzania, Zambia, and Zimbabwe.
- Implementation of ICH Q12 is in progress among ICH members and observers.
- Guidelines on submission of documentation for a multisource (generic) finished pharmaceutical product for the WHO Prequalification of Medicines Programme: quality part and quality information summary (QIS) guidelines on submission of documentation for a multisource (generic) finished pharmaceutical product for the WHO Prequalification of Medicines Programme: quality part.
- Several markets have developed unique national guidance and templates based on the WHO guidance to require the submission of a WHO QOS and/or QIS template in the initial marketing application. However, these countries may vary with respect to whether they consider these documents as commitments that must be maintained/updated with each subsequent variation.
- There is no official English version of the MPIS template available on the CDE/NMPA website.
- No specific regulation or guidance exists on the requirements for Republic of Korea CMC Summary; however, based on authors’ experience and regulatory queries we have included in our evaluation. The reference is to a parent guidance.
| SUMMARY DOCUMENT REQUIREMENT CTD SECTION |
WHO | CANADA | CHINA | JAPAN | REPUBLIC OF KOREA |
SOUTH AFRICA |
EAEU | PROPOSED1 |
| S11 NOMENCLATURE | Y | N | N | Y | Y | Y | Y | Y |
| S12 STRUCTURE | Y | Y | N | Y | Y | Y | Y | Y |
| S13 GENERAL PROPERTIES | Y | Y | N | N | N | Y | N | N |
| S21 MANUFACTURER | Y | Y | N | Y | N | Y | N | Y |
| S22 MANUFACTURING PROCESS | Y | Y | N | Y | N | Y | N | Y |
| S23 CONTROL OF MATERIALS | Y | N | N | Y | N | Y | N | Y |
| S24 CONTROL OF CRITICAL STEPS & INTERMEDIATES | Y | N | N | Y | N | N | N | Y |
| S25 PROCESS VALIDATION & EVALUATION | Y | N | N | N | N | N | N | N |
| S26 MANUFACTURING PROCESS DEVELOPMENT | Y | N | N | N | N | N | N | N |
| S31 ELUCIDATION OF STRUCTURE | Y | N | N | N | N | Y | N | N |
| S32 IMPURITIES | Y | Y | N | N | N | Y | N | N |
| S41 SPECIFICATION | Y | Y | Y | Y | Y | Y | N | Y |
| S42 ANALYTICAL PROCEDURES | Y | N | Y | Y | Y | N | N | Y |
| S43 VALIDATION OF ANALYTICAL PROCEDURES | Y | N | N | N | N | Y | N | Y |
| S44 BATCH ANALYSES | Y | N | N | N | N | Y | N | N |
| S45 JUSTIFICATION OF SPECIFICATION | Y | N | N | N | N | N | N | N |
| S5 REFERENCE STANDARDS OR MATERIALS | Y | N | N | Y | N | Y | N | Y |
| S6 CONTAINER CLOSURE SYSTEM | Y | Y | N | Y | N | Y | N | Y |
| S71 STABILITY | Y | Y | N | Y | N | Y | N | Y |
| S72 POST APPROVAL STABILITY PROTOCOL & COMMITMENT | Y | N | N | Y | N | N | N | N |
| S73 STABILITY DATA | Y | N | N | N | N | N | N | N |
| P1 DESCRIPTION AND COMPOSITION | Y | Y | Y | Y | Y | Y | Y | Y |
| P2 PHARMACEUTICAL DEVELOPMENT | Y | N | N | N | N | Y | N | N |
| P31 MANUFACTURERS | Y | Y | N | Y | N | Y | Y | Y |
| P32 BATCH FORMULA | Y | Y | N | Y | N | Y | N | Y |
| P33 MANUFACTURING PROCESS & CONTROLS | Y | Y | Y | Y | Y | Y | N | Y |
| P34 CONTROL OF CRITICAL STEPS & INTERMEDIATES | Y | Y | Y | Y | N | Y | N | Y |
| P35 PROCESS VALIDATION | Y | Y | N | N | N | Y | N | N |
| P41 EXCIPIENT SPECIFICATION | Y | N | Y | Y | Y | N | N | Y |
| P42 EXCIPIENT ANALYTICAL PROCEDURES | Y | N | Y | Y | Y | N | N | Y |
| P43 EXCIPIENT VALIDATION OF ANALYTICAL PROCEDURES | Y | N | Y | N | N | N | N | Y2 |
| P44 EXCIPIENT JUSTIFICATION OF SPECIFICATION | Y | N | N | N | N | N | N | N |
| P45 EXCIPIENTS OF HUMAN OR ANIMAL ORIGIN | Y | N | N | Y | N | N | N | Y |
| P46 NOVEL EXCIPIENTS | Y | N | N | N | N | N | N | Y |
| P51 SPECIFICATION | Y | Y | Y | Y | Y | Y | Y | Y |
| P52 ANALYTICAL PROCEDURES | Y | N | Y | Y | N | N | Y | Y |
| P53 VALIDATION OF ANALYTICAL PROCEDURES | Y | N | N | N | N | Y | N | Y2 |
| P54 BATCH ANALYSES | Y | N | N | N | N | Y | N | N |
| P55 CHARACTERIZATION OF IMPURITIES | Y | N | N | N | N | N | N | N |
| P56 JUSTIFICATION OF SPECIFICATION | Y | N | N | N | N | N | N | N |
| P6 REFERENCE STANDARDS OR MATERIALS | Y | N | N | Y | N | Y | N | Y |
| P7 CONTAINER CLOSURE SYSTEM | Y | Y | Y | Y | Y | Y | Y | Y |
| P81 STABILITY SUMMARY AND CONCLUSIONS | Y | Y | N | Y | Y | Y | Y | Y |
| P82 POST APPROVAL STABILITY PROTOCOL & COMMITMENT | Y | Y | N | Y | N | Y | N | N3 |
| P83 STABILITY DATA | Y | Y | N | N | N | Y | N | N |
- Proposal for a global summary document based on the ICH Q12 guideline on ECs.
- If performance-based ECs for analytical procedures are included in the application, then elements of the validation would need to be included as an EC.
- CMC regulatory commitments (e.g., stability, postapproval CMC commitment) made by a marketing authorization holder (MAH) to provide data or information to the regulatory agency in a market authorization application is considered supportive information.
CMC Documentation Requirements Beyond Content in M3
In addition to summaries of the “standard” quality content noted in Table 2, many of these compliance summary documents also require additional information above and beyond the ICH M3 requirements. This “extra” information may require life-cycle management. Table 3 summarizes these additional requirements for the markets within the scope of this article.
For example, the EAEU QND requires details of the drug product analytical methods to facilitate local testing. The Canada CPID, the South Africa SCoRE, and the WHO QOS require the drug product master batch record numbers, which are available at the manufacturing site upon inspection. These three compliance summary documents also include entries for the drug product process validation (PV) report and protocol document numbers, but this requirement could be addressed by providing the PV protocols and/or reports for review upon request.
The China MPIS has several unique requirements beyond the standard ICH M3 content, including that the excipient and packaging manufacturers and the China Drug Master file (DMF) numbers be listed, as well as a list of drug product manufacturing equipment. Notably, also required is the compendia version to which each compendial excipient complies, when it is broadly understood that each compendial excipient will comply with the compendia current at the time of the excipient’s use, without the need to update the drug product application. In addition to the MPIS, China also requires a specification/method document known as the “JX” for small molecules, which can also be considered a summary compliance document since it is a summary of M3 information that is required to be maintained after approval, but this document has been omitted for the purposes of this paper as its intent is for import testing.
Discussion
Regulators need a comprehensive, coherent description of the product control strategy [7] and an understanding of life-cycle change management, while industry needs to manage inconsistency in regulatory assessments and regulatory commitments for the same global product.
| Document Requirement | Canada | China | Japan | Republic of Korea |
South Africa | EAEU | WHO1 | Proposed |
|---|---|---|---|---|---|---|---|---|
| Detailed drug product (DP) analytical methods | X | |||||||
| Packaging process information | X | |||||||
| DP master batch record numbers | X | X | X | |||||
| Storage conditions for non-compendial excipients | X | |||||||
| Excipient manufacturers | X | |||||||
| Excipient registration numbers (China DMF) | X | |||||||
| Packaging component manufacturers | X | |||||||
| Packaging registration numbers (China DMF) | X | |||||||
| DP equipment details | X | X | ||||||
| Excipient compendial reference (including version) | X | |||||||
| DP in-use period | ||||||||
| Name/address of corporate headquarters | X | |||||||
| Name, position, education, and signature of person in charge at all sites listed in 3.2.P.3.1, as well as statement of commitment |
||||||||
| Quality certificate for each site listed in 3.2.P.3.1 | X | |||||||
| Japan Module 1.13 Tables 1 and 2 with details of parameters and acceptable ranges | X | |||||||
| PV protocol or report number | X | X | X | |||||
| Specific drug substance aqueous solubility | X | X | ||||||
| Drug substance specification number at DP manufacture site | X | X | X | |||||
| DP specification number at DP manufacture site | X | X | X |
- Requirements for content of WHO QOS may vary per market.
Challenge
While the QOS has been effective in certain ways, its ineffectiveness has led to requirements to provide additional summary documents (e.g., Canada CPID, China MPIS, Japan AF, Republic of Korea Summary document, South Africa SCoRE, and EAEU QND) beyond the QOS. These countries currently require a market authorization holder to provide specific summary documents as part of the regulatory submission and approval process for a new drug application.
In some cases, these summary documents contain the same information that is provided in the QOS sections in M2 and are therefore additional administrative tasks, which can create delays in submission and subsequent approval. In other cases, these documents require CMC information beyond what is required in the QOS and M3. For all of the countries discussed here, these additional summary documents represent commitments and must be maintained throughout a product’s life cycle, above and beyond the M3 documents. The provision and maintenance of additional market-specific summary documents is not conducive to supporting a harmonized QOS and presents an additional burden on industry.
Proposed Solution
Given that the purpose of these market-specific summary documents (with the exception of the EAEU QND) is to facilitate review of both the initial marketing application and subsequent post-approval changes, a single summary document could replace the QOS as well as each of these individual summaries.
Such a summary document has the potential to be an even more effective tool to significantly improve the efficiency and quality of the regulatory assessment. To do this, it needs to (a) efficiently describe the product development process in the context of mitigation of risk to the patient; (b) provide a clear summary of the overall control strategy as part of the risk mitigation or control; (c) guide the regulator through the fragmented content to the submission;7 and (d) elucidate the life cycle management plan for the product.
An opportunity exists to propose a summary document that does the following:
- Frames the drug development story by effectively conveying how enhanced process understanding, product knowledge, and risk assessments are linked to a comprehensive control strategy
- Links the drug substance and drug product critical quality attributes (CQAs) to target product profile (TPP) and quality target product profile (QTPP)
- Summarizes the holistic control strategy, including links to more detail in M3, demonstrating how the proposed manufacturing process and controls (namely, critical process parameters, critical material attributes, and ECs) will provide assurance a drug substance and drug product will meet their respective CQAs
- Declares and documents a summary of the ECs and a proposed PLCM, if applicable, can also be leveraged
A globally harmonized summary document that defines regulatory commitments, justified within a more comprehensive QOS document fulfilling global regulatory requirements, would benefit the regulators, industry, and most importantly, the patients. A comprehensive QOS that summarizes the product development in a narrative that connects the material attributes, process, and product understanding to the overall control strategy could improve the efficiency and economy of regulatory review and assessment of an application globally. In addition, the option leverages tools in the ICH Q12 guideline on “Technical and Regulatory Considerations for Pharmaceutical Product Lifecycle Management”14 in a single format that could provide regulators with a concise, risk-based submission summary highlighting a product’s control and life-cycle strategy, decreasing review and approval timelines, and enabling faster availability and sustained supply of critical medicines to patients worldwide.
Conclusion
The provision of market-specific compliance summary documents as part of the regulatory submission and approval process for a new drug application is considered beyond ICH requirements. In addition, effectively conveying enhanced process understanding and product knowledge in a regulatory application has been a challenge for both regulators and industry. A globally harmonized QOS in a regulatory submission that contains a comprehensive summary of the product development and control strategy, clearly listing all CMC commitments and how changes to them will be managed throughout the life cycle of a product, can address both of these challenges and would benefit regulators, industry, and patients by:
- Providing a relevant and risk-based summary of the entire CMC section of the marketing application and accelerating the regulatory action and decision-making process
- Facilitating approval of drug applications and improving the speed of patients’ access to new drugs
- Providing a succinct summary of the data to justify and confirm that the ECs are adequate to ensure product quality, building confidence in a product’s overall control strategy
- Clearly identifying the CMC regulatory commitments and eliminating the need for country-specific summary documents
- Leveraging the concepts and tools outlined in ICH Q12 to facilitate/simplify the management of post-approval CMC changes and enable continuous improvement and supply
The ICH Assembly finalized and released training modules on the ICH Q12 Guideline on Regulatory and Technical Considerations for Pharmaceutical Product Lifecycle Management on 10 June 2021. In addition, the 2020 ICH Assembly supported work on new topic proposals of CTD and Structured Product Quality Submissions, which is underway.8
Therefore, the timing is perfect to address the current challenges with the implementation of ICH M4Q (M2) expressed in this paper and to develop a globally harmonized QOS that benefits regulators, industry, and patients and eliminates the provision and maintenance of additional market-specific summary documents.
About the Authors




Acknowledgements
The authors wish to acknowledge the following for their assistance in the preparation of this article: Jacqueline Acquah, Phil Clements, Peilan Ding, Al-fonsus Karli, Megan McMahon, Satoshi Ohtake, Louise Akeyo Ongare, Margarita Polishchuk, Mike Saleh, Hiromi Sugano, Jun Yamada, and Marianne Zenon.


