A Patient-Centric Review of ICH Q6A and ICH6B

The ICH Quality Discussion Group (QDG) has recommended contemporizing the specification setting guidelines ICH Q6A and Q6B. This provides an excellent opportunity to update ICH Q6A/B to align and work in a complementary manner with the science-, risk/knowledge-based concepts in ICH Q8–Q12. The ISPE Pharmaceutical Quality Lifecycle Implementation (PQLI) Patient-Centric Specification working group has reviewed the current guidelines on how to improve the specification setting guidance. Recommendations are provided within this article.
The ICH guidelines Q6A “Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug Products” and Q6B “Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products” were first established in 1999.1, 2 Since then, they have served as a cornerstone for innovative pharmaceutical companies when establishing quality specifications for new products. These guidelines have enabled a harmonized approach to specification settings across the industry and have, to a certain degree, resulted in a convergence in regulatory authority application reviews.
A Patient-Centric Approach to Specification Setting
The science and risk/knowledge-based concepts outlined in ICH Q8–Q123, 4, 5, 6, 7 and impurity management guidance in ICH Q3D8 and ICH M79 encourage a holistic approach to developing the product control strategy by establishing relationships between process parameters and product quality attributes.
Under this paradigm, control is built into the manufacturing process, and drug substance and drug product specifications provide confirmation of product quality by means of testing select quality attributes as opposed to serving as the main basis of control. Based on this, the approach to setting quality specifications for a new drug substance (DS) and drug product (DP) should be reconsidered to reflect patient-centric requirements, focusing on developing specifications based on patient safety, product efficacy, and ensuring product supply, whereas process robustness and consistency are ensured via the application of an overall control strategy and continued process verification.
In this article, we recommend that DS and DP specification acceptance criteria be set considering the control strategy, which uses process and product understanding to link critical process parameters to critical quality attributes (CQAs). Particular attention is paid to CQAs associated with patient safety and, when relevant, product efficacy through the product shelf life and patient use.
This patient-centric approach to specification setting is frequently discussed, and the potential benefits throughout development and the product life cycle are well documented.10, 11, 12, 13 The core principles of patient-centric specifications apply to both synthetic molecules and biologics in that product control starts with foundational understanding of the intended product presentation, strength, and desired shelf life. This information is typically documented in target product profiles (TPPs) and quality target product profiles (QTPPs).
As this information is being developed, quality attributes are identified, and the level of risk to the patient of each is assessed. The risk management exercise identifies the level of impact on patients of each identified attribute, resulting in identification of CQAs. Foundational to our proposal is that not all CQAs need be included in a DS or DP specification, provided the control strategy provides adequate control of the attribute via other means.
For both synthetic molecules and biologics, control strategies ensure alignment with a product’s QTPP, with specifications being one of several possible control strategy elements that can be leveraged to confirm appropriate product quality. Prior knowledge and product understanding can also be important inputs to a patient-centric control strategy.14 All CQAs should be controlled through a product’s control strategy, but it is important to note that not all CQAs need to be included in the final product specification.
Inclusion or exclusion of an attribute and associated analytical procedure on a product specification should primarily depend on the level of risk of the attribute (i.e., impact to patient safety and efficacy, also often referred to as criticality); the ability of the process to demonstrate robust control of the attribute as demonstrated through appropriate scientific studies; and confirmation of attribute control through in-process testing, validation, and verification. CQAs whose control was established by other robustly justified means do not need to be included in the specification.
Synthetic molecules and biologic molecules diverge in the specification setting process in some of the nuances and regulatory expectations associated with specifications. However, for both synthetic and biologic molecules, the justification of acceptance criteria beyond historical experience can be established by leveraging quality risk management principles, data safety and toxicology, process development, immunogenicity, pharmacokinetics (PK), and potency information (published or internal) from similar molecules or from the molecule of interest.
Although qualified levels of impurities are a major focus of synthetic molecules, defined and qualified impurity levels that would be acceptable for biologics are rarely available. Potency determination by some form of bioassay is a requirement for biologic molecules, in addition to strength and content (as opposed to a single “assay” requirement for synthetic molecules), and potency assays are complex and far more variable than many analytical procedures applied to synthetic molecules. Only select contaminants potentially found in biological products have levels governed by regulatory guidance. Other impurities and attributes in biologic molecules rely on holistic control strategies that take process and product understanding into account.
Comparisons between typical specification components for synthetic molecules and biologics, based on published information,11, 15 may be found in Tables 1 and 2. For the DP comparison, the solid oral dosage form is compared to an injectable biologics product because these are the most common dosage forms for these product types.
| Synthetic Molecules | Biologics | Comments |
|---|---|---|
| Description | Appearance | |
| Identity | Identity | |
| Identity: chiral identity | ||
| Counterion | Biologics are not isolated as salts | |
| Assay | Quantity or strength | |
| Potency | Potency is measured by bioassays (e.g., ELISA, cell-based assays, or in vivo bioassays), which have significant inherent variability | |
| pH | Included for biologics to ensure consistent and appropriate formulation | |
| Impurities and degradation products | Purity and product variants | For biologics, there are usually multiple separate tests for measuring purity and product variants |
| Chiral impurities | ||
| Residual solvents | Process-related impurities | Additionally for biologics, many process-related synthetic molecule impurities are evaluated during process validation and are often not included in the specification |
| Inorganic impurities | For biologics, elemental impurities are evaluated during DP process development/validation | |
| Water content | ||
| Polymorphic forms | ||
| Particle size | ||
| Microbiological attributes | For biologics, endotoxin and bioburden are required |
| Synthetic Molecules | Biologics | Comments |
|---|---|---|
| Description | Appearance and description | |
| Identity | Identity | |
| Assay | Quantity or strength | |
| Potency | Potency is measured by bioassays (e.g., ELISA, cell-based assays or in vivo bioassays), which have significant inherent variability. | |
| pH | Included for biologics to ensure consistent and appropriate formulation | |
| Osmolality | Included for biologics to ensure consistent and appropriate formulation | |
| Reconstitution time (for lyophilized products) | ||
| Water content | Moisture content (for lyophilized products) | |
| Extractable volume | ||
| Excipients | For biologics, select excipients can be CQAs; synthetic molecules may include this attribute | |
| Degradation products | Purity and product variants | For biologics, there are usually multiple separate tests for measuring purity and product variants |
| Uniformity of dosage units | Uniformity of dosage units can also be a requirement for lyophilized biologics | |
| Dissolution | ||
| Microbiological attributes | For biologics, endotoxin and sterility are required |
The intention of this article is to emphasize those aspects of ICH Q6A and ICH Q6B that embrace the patient-centric, science-, and risk-based approach to specification setting and identify opportunities for those that do not. It may not be possible or appropriate to have a “one-size-fits-all” approach to specification setting. The approach for establishing acceptance criteria will be linked to the process development knowledge established and clearly presented in an application by the applicant.
Enhanced development may provide a larger set of knowledge to be used in justification of the proposed control strategy. Process and product development as well as understanding of and knowledge for any given product will differ, even within a company, based on the speed and stage of development, availability of platform approaches, complexity of process, and other factors. The decision trees proposed in this article may be applied regardless of development strategy.
Recommendations
We conducted a comprehensive review of ICH Q6A and reviewed each decision tree in detail (see Table 3). Many of the existing decision trees already incorporate and prioritize a patient-centric approach to set specification limits. The recommendations herein focus on the decision trees that prioritize other considerations, such as batch history and the limited manufacturing experience at the time of commercial application submission.
| Decision Tree | Assessment of Patient Centricity |
|---|---|
| #1 Establishing acceptance criterion for a specified impurity in a drug substance | The primary consideration driving the limit is a statistical assessment of the batch data. An alternate approach is provided and discussed with Figure 2. |
| #2 Establishing acceptance criterion for a degradation product in a new drug product | The decision tree recommends limiting levels to those demonstrated within the batch history. A simplified approach is discussed within this paper. |
| #3 Setting acceptance criteria for drug substance particle size distribution | The decision tree aligns with the principles of patient centricity in that it considers the impact of particle size on patient relevant attributes, such as bioavailability, dissolution, and content uniformity. |
| #4 Investigating the need to set acceptance criteria for polymorphism in drug substances and drug products | The decision tree focuses on how polymorph formation might affect patient-centric properties, efficacy, and safety. |
| #5 Establishing identity, assay and enantiomeric impurity procedures for chiral new drug substances and new drug products containing chiral drug substances | The decision tree links the need for testing/limits to the sponsor’s knowledge base, with respect to whether racemization can occur after chiral purity is established. |
| #6 Microbiological quality attributes of drug substance and excipients | The need for testing/limits is linked to product understanding and prior knowledge. |
| #7 Setting acceptance criteria for drug product dissolution | The decision tree links criteria requirements to the Biopharmaceutics Classification System (BCS) characteristics to the drug, focusing on patient impact. |
| #8 Microbiological attributes of non-sterile drug products | The decision tree focuses on the knowledge and risk the drug can support microbial growth and the recommendations align with product understanding. |
Development strategies that adopt the principles of enhanced development outlined within ICH Q8–Q11 may develop an understanding of the capability of a manufacturing process well beyond the results provided in a product’s batch history (e.g., through fate/purge experiments, multivariate process understanding studies, and/or structure function studies). The proposed decision trees (see Figures 1–4) embrace this process and product understanding as part of the specification limit setting process. Setting limits in this manner, rather than according to batch history at the time of regulatory submission of a commercial application, also enables continual improvement in a holistic way, as ICH Q12 encourages.
Decision Tree for Removal of an Analytical Test
Currently, ICH Q6A/B does not discuss when a quality attribute requires control (e.g., process or analytical controls). These concepts are addressed within other ICH guidelines for specific attributes, such as ICH M7 for mutagenic impurities and ICH Q3D for elemental impurities. Often, omission of a test may be justified when the levels for the attribute of concern are reliably controlled by the manufacturing process and/or upstream analytical controls. A decision tree that clearly articulates the expectations for such a justification could enable further regulatory harmonization for acceptance of such a control strategy.
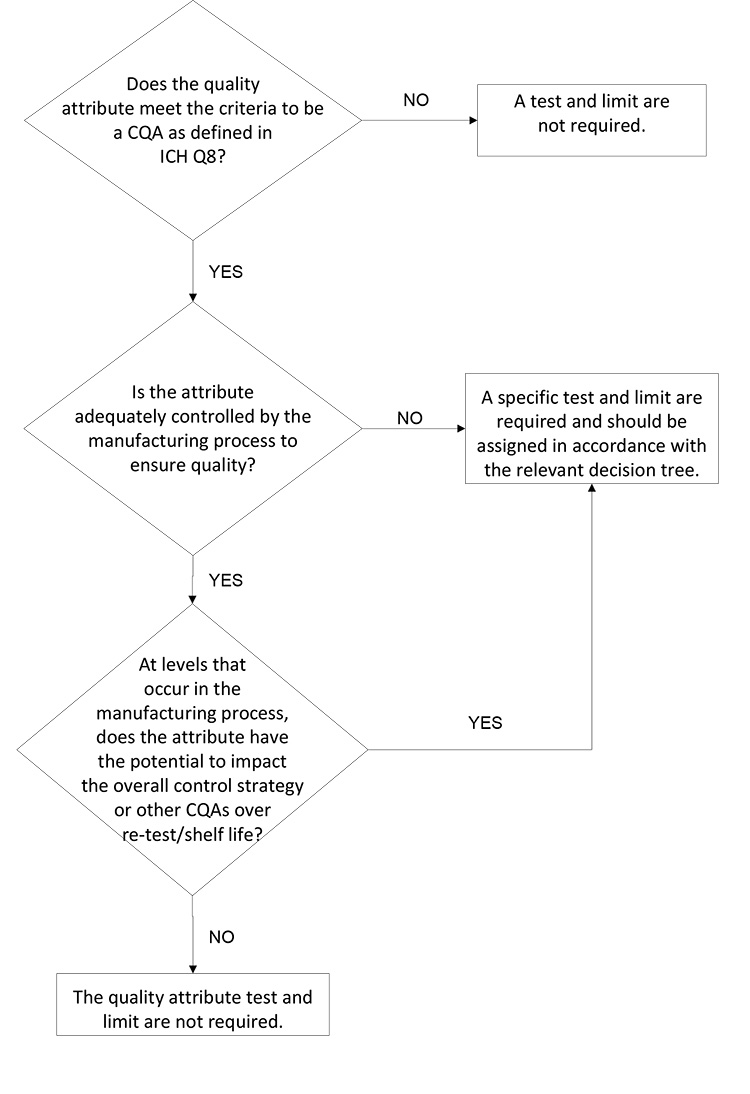
A proposed decision tree for assessing whether an attribute should be included on the DS or DP specification is presented in Figure 1. This decision tree could be applied to both synthetic and biologic molecules; A biologic molecule version of this decision tree is provided and discussed in Figure 4 and considers biologic molecule modality differences not considered in Figure 1.
The first decision box in Figure 1 prompts the company to consider whether the attribute in question is a CQA. If the attribute is not a CQA, specified control is not required. If the attribute is a CQA, it may be included on a DS or DP specification, it may be confirmed via an in-process control, or it may be demonstrated that no routine analytical confirmation is needed if robust process control can be demonstrated. For clarity, it may be useful to define what qualifies as “robust process control”; for example, ICH Q3D and ICH M7 define the impurity purge level expected to justify not testing for elemental or mutagenic impurity.
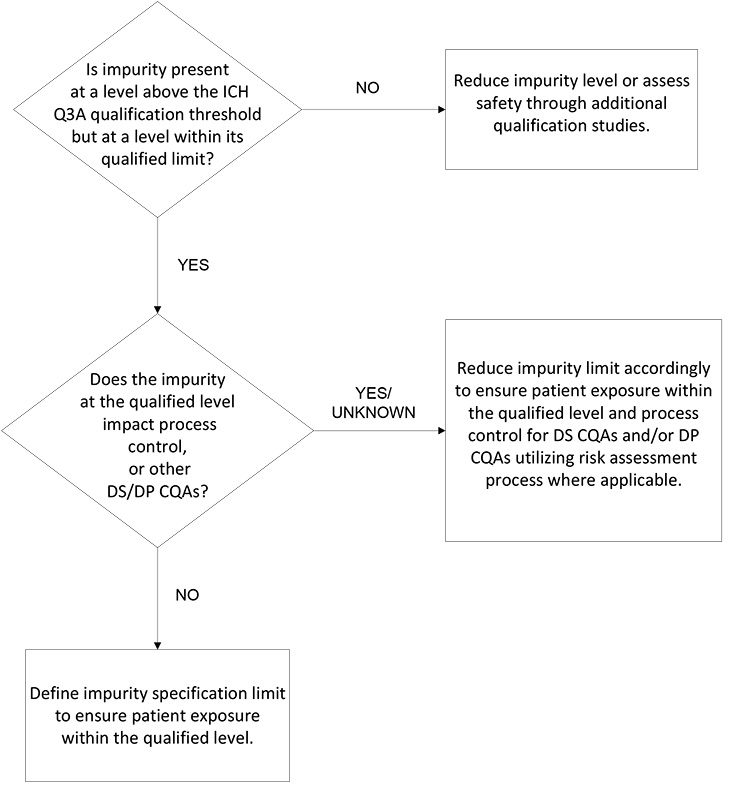
Synthetic Molecule DS Impurity Acceptance
In the 20 years since ICH Q6A was initially implemented, there has been an evolution of regulatory expectations based, in part, on improved technology, i.e., sensitivity of analytical methods, introduction of the science- and risk-based approach, and elaboration of the pharmaceutical quality system.
In this light, the synthetic molecule DS’s impurity acceptance criteria proposed by a sponsor should be designed in line with science- and risk-based principles rather than with batch data and a statistical approach. This includes a holistic approach that considers all development studies (e.g., toxicology studies, manufacturing process development, development and scale-up batch data, and evaluation of degradation associated with similar compounds).
The decision tree in Figure 2 is focused on patient safety and assurance of product quality. It considers the different inputs such that the sponsor is focused on an impurity control approach with patient safety, supply, and life cycle in mind. This approach assures the impurity level is within demonstrated safe levels and that the impurity will not compromise other quality attributes or process controls (i.e., any component of the overall control strategy). If development data supports that an impurity present at the qualified level per ICH Q3A will not negatively impact the manufacturing process control strategy or other CQAs, then the qualified level serves as an appropriate limit for the impurity.
Synthetic Molecule DP Degradation Product Limits
For the DP, a limit for each degradation product may solely consider the impurity’s impact on patient safety and efficacy and consider potential changes over the shelf life and patient-use period. If there are no concerns about impact on product safety and efficacy, the impurity limit may be set based on the qualified level and considering any degradation. If the degradation product could impact patient safety or product efficacy, then limits should consider the appropriate level at the end of shelf life (EOSL) and an in-use period, if applicable.
If a DP contains several degradation products with qualified levels above the unspecified limit, overall product quality should be considered in addition to the safe level of each individual degradation product. The applicant must establish limits for each degradation product while also considering the impact to other acceptance criteria, such as product potency and the acceptable total degradation product limit. A decision tree is not proposed for the degradation products in synthetic molecule DPs, due to the simplicity of the proposed strategy.
Water Content for Synthetic Molecule DP
Per ICH Q6A, a test for water content in the DP should be included when appropriate. However, currently, ICH Q6A does not include a decision tree for testing water content in the DP. In many cases, water content in a synthetic-molecule, solid dosage form DP would not adversely impact the product’s safety, quality, or efficacy. Any CQAs that could potentially be impacted by water content, such as assay and degradation products, appearance, dissolution, and physical form or microbial limit in the DP, are typically tested directly.
The decision tree in Figure 3 is proposed based on understanding of potential risk and impact of water content to product safety and quality, with the intent to provide guidance and consistency around setting any necessary and appropriate water content control in DP.
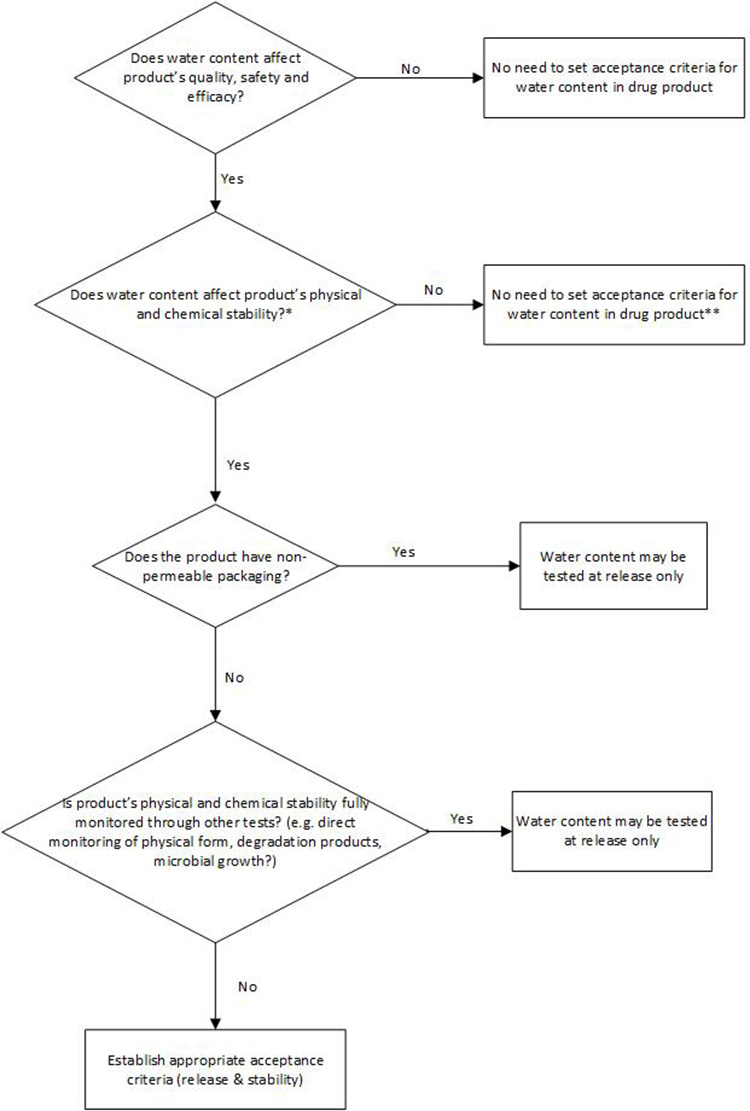
** Alternative control should be in place, e.g., process control
Specification Considerations for Biologics
Some of the recommendations described previously are also relevant for biologics. However, the complexity of biologics often results in several product-related variants, some of which may be critical to the product’s safety and efficacy and some of which may be non-critical. Complex bioprocesses can result in biologics that consist of a mixture of post-translationally modified forms, many of which can increase throughout the product’s shelf life. Product-related variants in biologics can include, but are not limited to, glycosylation and N/C terminal processing variants, oxidation, deamidation, isomerized, aggregation, fragmentation, glycation, and disulfide bond breakage or scrambling products, and additional post-translational modifications not mentioned previously.
Historically, ICH Q6B has used the term “product-related substances” to refer to molecular variants that have comparable activity, efficacy, and safety relative to those of the desired product (i.e., variants that are not critical), with acceptance criteria for product-related substances set either individually or collectively. More recently, however, the “product-related substances” terminology has largely been supplanted by discussion about attribute criticality and how it ties into the quality risk management for a given product.
In line with and considering quality risk management principles outlined in ICH Q8–Q11, a change in terminology used in ICH Q6 is recommended. For product-related substances that do not impact safety or efficacy and are thus not CQAs, a routine release test with corresponding acceptance criteria should not be required.
If an attribute that does not impact safety or efficacy is considered important for assessment of process efficiency, it would be appropriate to manage within the company’s pharmaceutical quality system (PQS), e.g., through internal limits, as opposed to implementing registered specification limits.
CQAs are required to be controlled via a control strategy which may include process, analytical, or other controls.15 While CQAs often have tests with acceptance criteria, there can be situations where CQAs are fully controlled by process controls. For example, DNA and host cell proteins are CQAs that can often be excluded from a commercial release specification due to consistent demonstrated removal during development and process validation batches.
Furthermore, some test methods may quantitate a combination of CQAs and non-critical quality attributes (non-CQAs) based on the overall product-related variant profile, in which case the acceptance criteria can take into consideration the attributes and their criticality to either focus the acceptance criteria on only the CQAs or consider more flexible acceptance criteria if the reportable value is a summed result that includes both critical and non-critical attributes.
Product-related impurities are variants that do not have comparable activity, efficacy, and/or a safety profile relative to the desired product and are thus CQAs. Historically, acceptance criteria for product-related impurities can be set either individually or collectively and should be based on data obtained from batches used in preclinical and clinical studies and manufacturing consistency batches.
The patient-centric approach is not limited solely to batch data but also takes prior knowledge and product knowledge under consideration. Quality risk management principles and the emergence of data which can enable justification beyond historical experience can be leveraged where justified.
Safety, immunogenicity, pharmacokinetic, and potency information (published or internal) from similar molecules or from the molecule of interest can be leveraged to support acceptance criteria that might be wider than batch data. Structure function studies and/or linkage of CQAs to clinical experience can also play an important role in selecting and justifying acceptance criteria. Furthermore, toxicology and clinical dose-ranging levels are relevant considerations.
For biologics, some impurities may not have “qualified levels” (i.e., qualified in toxicology studies) or enough other supporting information (e.g., prior knowledge, dose-ranging studies, structure function knowledge) to justify acceptance criteria in any way other than batch data. In cases where data from batches is used in preclinical or clinical studies, as well as actual manufacturing batches that need to be leveraged, considering appropriate statistical ranges to establish acceptance criteria around the available datasets can be useful.
Where limited clinical and batch history data is available, reliance on minimum, maximum, or exceedingly tight statistical ranges (e.g., three standard deviations as opposed to tolerance intervals) can lead to overly narrow acceptance criteria and hinder continued supply. In those cases, alignment on an initial criterion that enables product supply with a regulatory agreement (e.g., a follow-up measure) to further refine the acceptance criteria upon availability of sufficient batch data is advisable.
It should be noted that some biologics may exhibit change on stability requiring separate acceptance criteria for release and EOSL. Furthermore, when acceptance criteria are set based on batch history and stability data and a molecule exhibits additional changes during in-use stability, a third tier of acceptance criteria that takes in-use stability data into account may be necessary.
Acceptance criteria set based on long-term stability data alone are unlikely to account for further acceptable changes occurring under higher in-use temperatures and could result in unnecessarily tight acceptance criteria that prevent practical use of the DP.
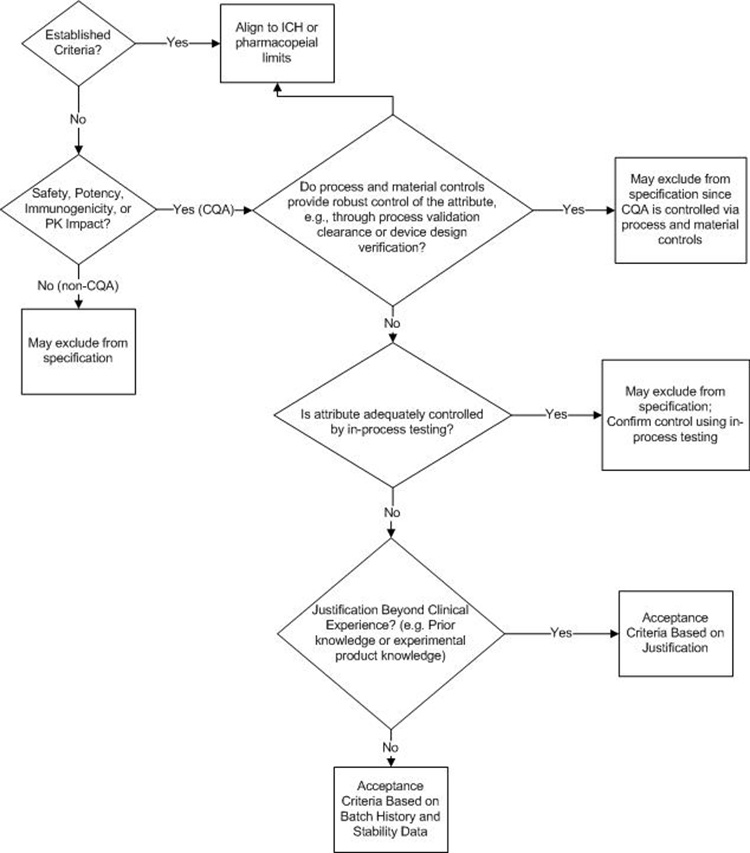
The decision tree presented in Figure 4 provides guidance around setting specifications for biologics. Specific elements to note include the following:
- Quality attribute criticality, with particular focus on safety, potency, immunogenicity, and/or pharmacokinetics, is the foundation for establishing the need for a specification attribute and the associated acceptance criteria.
- Alignment to ICH specification guidance and/or major market compendial standards.
- For attributes that are not easily defined by compendial requirements, prior knowledge, or qualified levels, applying a statistical evaluation of relevant batches—including the use of valid statistical approaches to aid in defining acceptance criteria—can define a baseline for acceptance criteria.
- Use of risk-based justification based on scientific and prior knowledge to broaden beyond clinical exposure.15
- If limited product knowledge and batch data are available, other approaches—including filing an initial specification to be updated after a requisite number of batches are manufactured—can and should be considered.
Discussion
Since the introduction of ICH Q6A and Q6B in 1999, the pharmaceutical industry has experienced several trends that have increased the need to move toward a more science- and risk-based approach to setting acceptance criteria. An increased focus on both targeting niche indications in smaller patient populations and more accelerated clinical programs may potentially lead to a reduction in the number of batches of DS and DP needed during the development phase.
This reduces the availability of sufficiently large batch data sets at clinical and commercial scale and at the commercial site, and even using the commercial process. Furthermore, the challenge of delivering new medicines to meet unmet medical needs has driven the adoption of accelerated chemistry, manufacturing, and controls (CMC) development programs, utilizing fast-track/early access initiatives offered by many health authorities (e.g., PRIME by the European Medicines Agency and the Fast Track program, breakthrough therapy, and accelerated approval by the FDA).
The trend over the last 20 years has, therefore, been toward fewer batches at commercial scale being available at time of registration. Although this situation is briefly acknowledged in ICH Q6A and there is a clear statement that where limited batch data is available at time of registration, the basis for specification acceptance criteria should focus on safety and efficacy, the lack of sufficient batch history is now commonplace and should be treated as such. In most circumstances, the sponsor will not have a data set large enough to convey process knowledge and understanding. Rather, this understanding is gained and demonstrated through development work.
Overreliance on batch history for setting specification limits at the time of registration may result in limits that are too tight if, as is often typical, the manufacturing experience at the time of registration is too limited to accurately describe the process variability expected through a product’s life. In turn, this may result in unnecessary product rejection, shortage of products to patients, and increased manufacturing costs. With typically limited availability of batch data at time of filing, incorporating process consistency considerations into specification acceptance criteria should be avoided in preference for wider patient-centric considerations that focus on safety and DP efficacy.
Assurance that the process remains in a state of control (the validated state) during commercial manufacture is best managed through continued process verification 1 within an appropriate quality management system that assures adherence to cGMP requirements. Here, an adaptive, continued process verification program will plan postapproval data monitoring, statistical evaluation, and trending that adjusts over a process's life cycle and will detect and appropriately act upon process variability. The data generated by the continued process verification program would also provide ongoing support of the validity of the commercial specification.16
Many pharmaceutical companies have sustainability targets associated with the development of new processes. These can be negatively impacted by unnecessarily tight specifications. For example, high solvent volumes are often used to wash materials to decrease the quantity of impurity from levels that are safe and do not impact DP efficacy to levels close to zero. Other examples include overly tight acceptance limits impacting supply chain robustness, such as the reprocessing of safe and efficacious material to meet specifications based on batch data of small number of lots made in development. Equally, unnecessarily tight specifications can reduce the permissible shelf life of medicine or mandate unnecessary cold storage supply chains.
The concepts of evaluating acceptance criteria considering safety data (permitted daily exposure [PDE]) and impact on CQAs is already established within ICH guidance, specifically ICH Q3D (R1), where it is stated that “[the] applicant is not expected to tighten the limits based on process capability, provided that the elemental impurities in drug products do not exceed the PDEs [...] In some cases, lower levels of elemental impurities may be warranted when levels below toxicity thresholds have been shown to have an impact on other quality attributes of the drug product.”
As written, the text within ICH Q6A clearly acknowledges that at the time of submission, available batch data is limited and it may be inappropriate to set criteria on batch data alone. However, ICH Q6A decision tree #1 is frequently interpreted differently because the first decision point is a definition of the range observed to date in the batch data. The result of this is unharmonized interpretation by regulatory reviewers.
Although some regions embrace an approach based on safety and product understanding, others still point to batch data as the primary source for the justification of the specification limits. Ultimately, this leads to divergent regional specifications which, in turn, creates complex supply chains where batches are selected for release to specific regions, increasing manufacturing costs and potentially unnecessarily increasing waste.
Embracing a patient-centric approach to specification setting has potential benefits to health agencies, patients, and the pharmaceutical industry as follows.
Benefit to patients:
- Ensures continued commitment to ensure patient safety, efficacy, and overall product quality
- Minimizes potential for supply shortages
- Encourages life cycle efficiency improvements, minimizing cost and environmental impact
Benefits to health agencies:
- Provides a consistent and robust approach to assessing limits that ensure patient safety, efficacy, and product quality
- Promotes investment in robust development, comprehensive control strategies, and process understanding
- Encourages life cycle improvements, such as reduction of environmental impact
Benefits to industry:
- Enables limit setting that ensures patient safety, efficacy, and product quality
- Helps ensure supply needs can be met
- Enables life cycle changes
- Minimizes waste and batch failure
- Minimizes regulatory divergence, e.g., differing regional limits17
- Reduces unnecessary development work
ICH Q8–Q11 set the stage for moving toward a comprehensive control strategy, thereby removing reliance on end product testing to ensure product quality. The combination of robust process development and understanding with a strong quality risk management system provides manufacturing facilities with the tools they need to ensure robustness and product quality. Therefore, it is time to reconsider the value of batch history when establishing specifications. With a robust control strategy in place, the limits listed on the specification are ensured.
Furthermore, with the onset of ICH Q12 and its emphasis on enabling continual improvement (i.e., improving process efficiency and robustness), it is even more important to set specification limits with the product life cycle in mind. Doing this effectively correlates to enabling a robust product supply chain while also encouraging manufacturers to continually improve their manufacturing processes.
These improvements may translate to increased efficiency and robustness of the process. They also help ensure consistent global supply and may enable the adoption of new green technologies resulting in less waste and environmental impact. Furthermore, they are more attainable within the context of quality standards—i.e., a standard directly linked to ensuring patient safety and product efficacy rather than maintaining a quality standard defined by manufacturing experience at a point in time (e.g., at the time of submission).
Conclusion
A baseline approach to pharmaceutical development, as defined by ICH Q8 (Appendix 1), leads to a reliance on end product testing to ensure quality through product specifications that are defined primarily by the manufacturing history at the time of registration. With increasing adoption of enhanced quality by design approaches that keep the patient in mind, the primacy of batch data in specification setting should be reevaluated. Emphasis should be shifted to the utilization of process knowledge and understanding of the relevant factors and their impact on the desired product efficacy and safety.
The recommendations outlined in this article aim to promote strategies for specification setting that consider the patient-centric, holistic product control strategy. These recommendations reflect the spirit of ICH Q8–Q12 and would align ICH Q6A and Q6B more closely with these guidelines. Additionally, the recommendations and clarifications outlined within would further enable global acceptance of proposed product specification limits. Globally harmonized specification limits, in turn, would enable a company to apply robust life cycle improvements enabled by ICH Q12. These specification limits would also enable a robust supply chain and important efficiency and environmental improvements, positively impacting patients.
Acknowledgments
The authors would like to thank the following colleagues for their thoughtful reviews and constructive feedback: Marta Czupryn, Margaret Ruesch, Jonathan Beaman, Ron Ogilvie, Olivier Dirat, Timothy Watson, Joann Parker, Filippos Kesisoglou, Prasanna Reddy, Kathy Lee, Alexander Pavon, and Steven Nowak.


