Concluding Compliance Challenges with Validation 4.0

As the pharma industry moves to an ambitious Validation 4.0 paradigm, computerized systems play a pivotal role in enabling the rapid transition. Innovation and agility in computerized system validation (CSV) received a strong push in the second half of 2022 with the publication of the FDA draft guidance on “Computer Software Assurance for Production and Quality System Software”1 and the ISPE GAMP® 5 Guide: A Risk-Based Approach to Compliant GxP Computerized Systems (Second Edition)2.
Within ISPE, there has been further discussions on a model for Validation 4.0 as an integrated process with articles like “Industry Perspective: Validation 4.0 - Shifting Paradigms” [3]. Furthermore, there have been discussions on moving away from legacy maintenance of massive documentation to digital artifacts and tools to provide a real-time view of compliance and control [4]. This article aims to provide a high-level perspective on seven key pillars for a Validation 4.0 approach with reference to some subtle variance from the legacy CSV approach.
PROGRESS TOWARD VALIDATION 4.0
The methodology in pharmaceutical companies is evolving to keep pace with draft industry guidance and changing validation expectations, which introduces challenges and opportunities typical of any radical transformation. Information technology (IT) quality division, IT managers, and consultants who have been used to a rigorous documentation and testing approach are suddenly exposed to the possibility of a much more lenient way of working.
This has introduced questions about practical implementation and potential risks with the sudden downgrade. Business owners who, in the new approach, now have a greater onus for accurately defining the risks at a more granular level are also trying to adjust their understanding, availability, and collaboration approach to rise to the occasion.
| Key Pillars of Validation 4.0 |
|---|
| 1. Process and data flows as foundation for validation activities |
| 2. Digital-tool-based artifacts for system documentation |
| 3. Critical-thinking-based risk approach for assurance activities |
| 4. Optimizing test scripting rigor |
| 5. Automated and pragmatic test execution approach |
| 6. Integrating efforts with cybersecurity and other regulatory units |
| 7. Continuous control and cognitive compliance |
PROCESS AND DATA FLOWS FOR VALIDATION ACTIVITIES
Validation 4.0 envisages a validation model that incorporates the process and key data early in design to get a head start on defining the associated risk and needed controls [4]. There is also a possibility of completely replacing user requirements specifications. In the context of CSV, although traditional user requirements specifications are replaceable, individual user requirement statements linked to parent process or data flows may still be necessary as a unique source of traceability.
It is important to take process and data flows as a foundation for understanding the business process for definition of risks and all linked downstream activities. This is also key to understanding any data integrity flaws introduced with manual interventions and the need for additional control points in the process flow to mitigate these risks.
An important aspect here is also the possibility of deriving requirements directly from the process and data flow diagrams. Traditionally, these flow diagrams have been considered a purely business topic and have been created, maintained, and referred to by business organization only—with CSV activities starting only at the user requirements level. User requirements could be derived directly from data flow diagrams if marked appropriately as a supported system rather than duplicating the efforts to write user requirements again.
Using the actual process flows employed by business organizations as starting points for scaling CSV efforts also drives consistent process nomenclature and better collaboration between business and IT organizations. Some existing tools (e.g., business process model and notation) may already be supporting this functionality. Effort could be made to fine-tune it to efficiently derive requirements where possible from process and data flow diagrams or to link newly created requirements to flow diagrams entities.
DIGITAL-TOOL-BASED ARTIFACTS FOR SYSTEM DOCUMENTATION
There has been a lot of discussion around paperless validation, and pharma companies are in different stages of adaption. Industry-standard application life cycle management tools have come in handy. The move to agile ways of working has also accelerated this process because agile approaches necessitate the use of tools to manage the scrum deliverables.
Some items in the validation life cycle have been easily automated: for example, test management and execution and requirements management with multiple commercially available options. There are still practical challenges to integration or completely moving all validation deliverables to tools. This is due to the lack of capability of any single tool to provide flexibility, like Microsoft Office Suite products do, to produce diagrams, tables, annotations, flow charts, and technical details. It has been further complicated with the need for compliance with 21 CFR Part 11 requirements10.
An important constraint in innovation for these supporting tools has been the confusion around electronic signature/approval and data integrity requirements. Most industry-standard software tools supporting agile delivery are easy to use but have limited functionality for capturing multiple electronic signatures.
To meet the varying needs of diverse stakeholders, organizations must use the best product available for capturing a particular activity and collate and approve that information in a single repository for completion of validation process. This has also led to cases where the best tools are used for ease, but information is extracted and approved in another validated document management system, leading to overhead efforts. Usage of different tools also leads to different naming conventions and redundancies in managing traceability.
VALIDATION 4.0 GOALS
The Validation 4.0 model should aim to accomplish the following goals.
Encourage Use of Digital Documentation and Metadata
One of the key extrinsic values of using digital documentation is that it transforms information into knowledge quickly5. Extensive use of metadata (i.e., data about data) directly in the tool makes searching, filtering, and reporting easier. This metadata can include country and site codes, traceability, keywords, system impacted, change requests, etc.
Digital documentation also helps achieve principles for attributable, legible, contemporaneous, original, and accurate (ALCOA) with detailed audit trails. For example, a Microsoft Word–based document may need to enable track changes but would not have a track change history retained for all approved versions. However, a digital version can be configured to keep mandatory detailed change tracking starting from the first approved version.
Minimize Validation Expectation for Software Tools
As per GAMP® 5 Second Edition, “tools and systems supporting system life cycles, IT processes and infrastructure should not be subject to specific validation, but rather managed by routine company assessment and assurance practices and good IT practices” within industry standard frameworks like ITIL [Information Technology Infrastructure Library] as they do not directly support GxP-regulated business processes or GxP records and data2. To buttress this point, the GAMP guidance has categorized these tools as GAMP Category 1.
Furthermore, the full scale of data integrity controls and electronic signatures would also not apply. Regulated companies should explore how best to use a standard audit trail and simple approval functionalities of these tools to provide reasonable assurance of review and acceptance. Locking the documentation objects after approval and other version control aspects also may need to be considered.
Avoid Redundancy in Tool Usage
With reduced expectation for e-signature controls, the need for extracting requirements or testing done with best available tools, and reapproving in a validated document management system as consolidated documents, also becomes unnecessary. Reports and logs extracted from these tools can be used to achieve pragmatic reviews in a consolidated view. They can also be used as supporting documents for providing baseline status for requirements, test reports, traceability matrix, etc.
Employ a Holistic Document Management System
The system should fulfill the needs and flexibility of different stakeholders and documentation types. It should preferably use agile tools, a basic approval workflow, audit trails, and version control functionalities. These should be supported by some key dedicated modules covering:
- Business modeling that is customizable for easily defining business process flows and roles
- Requirement description (extractable/derived from business process modeling) and additional new individual requirements (nonfunctional, controls, etc.) with option to define requirement types
- An integrated functional specification, design, and configuration specification module with the potential to add interface description and flows, descriptive attachments, etc.
- Test scripting and execution, supporting easy defect management and reporting
- Validation and project management, offering flexibilities in line with Microsoft Word and Excel
CRITICAL-THINKING-BASED RISK APPROACH FOR ASSURANCE ACTIVITIES
The FDA draft guidance defines Computer Software Assurance (CSA) as “a risk-based approach for establishing and maintaining confidence that software is fit for its intended use” 1. Risk-based approaches have always been used in pharmaceutical IT, but they have primarily been a checklist activity, with the majority of the outcome focused on a scripted testing type for every GxP functionality. The new approach for Validation 4.0 should enable the shift in focus and effort for all stakeholders from testing to accurate risk assessments to determine holistic assurance activities with application of critical thinking.
The ISPE GAMP® Good Practice Guide: Enabling Innovation illustrates critical thinking as “proactive adoption of a risk-based approach suitable for the intended use of the computerized system that takes into account the multiple layers of assurance provided by the business process”6. Note that as part of a comprehensive assurance approach, and as described in GAMP® 5 Second Edition and ICH Q9, the primary objective of quality risk management is to identify and assess GxP risks2, 7. This is so that appropriate controls may be applied to protect product quality and patient safety, and not just to support the use of risk scoring to optimize testing approaches.
This shift needs to be more organic and requires a transition at different levels. Some transitions are outlined next.
Comprehensive Assurance Approach
The new approach would require understanding assurance as a full spectrum of activity, combining process, design, and manual controls with testing as a final step of assurance. Key examples include improving the process or system design, understanding four-eye and review checks by quality personnel in the business process, increasing the level of detail of specification, design reviews, etc.
Binary Risk Definition and Patient Safety Focus
An important enabler for all stakeholders in accurate assessment of risks would be to keep the definitions at a binary level only. Traditionally, regulated companies tend to categorize risks with multiple values for each risk, e.g., high, medium, low. This is problematic, especially for practitioners when trying to fit between these values with subjective judgements. Binary values can make decision-making easier and faster.
The FDA draft guidance noted the importance of outlining binary risks and limiting the definition of risks to only be based on the impact to patient safety. This is compared to the traditional approach of considering patient safety, product quality, and data integrity. The new approach also considers product quality as reference but ultimately checks how the risk to product quality impacts patient safety. A single-minded focus would help stakeholders clearly understand the impact.
Use Identification and Traceability at the Appropriate Level
As per the FDA draft guidance on CSA, software may have multiple intended uses depending on the features, functions, and operations1. It may also present different risks with different levels of validation efforts required. The current approach of functional risk assessment may not always be suitable to capture the level of details required to ringfence risks. The risk assurance approach should have an enabler, and a traceability approach can be a good fit.
There have been different approaches used to manage traceability—from a generic document level to an atomic level. A Validation 4.0 approach would need traceability at a more atomic level. That is, each user requirement would need to be linked to a unique functional requirement (that can represent a specific feature fulfilled by the system rather than a functionality), which is in turn linked to a unique test case (rather than a generic test script). This would also be an enabler for inheritance of risks, as described in Figure 1.
Risk Inheritance Concept
It can often become tedious to define GxP risk for every feature or requirement, and a concept of inheritance may be considered to optimize risk assignment. GAMP® 5 Second Edition encourages the use of a hierarchical approach to help simplify risk assessment, and it also hints at the use of major and subsidiary functions or requirements to group similar entities2.
A good starting point for this hierarchical approach can be a process risk assessment. As per GAMP® 5 Second Edition, “a process risk assessment (also known as business process risk assessment) is a non-system-specific high-level assessment of the business process or data flow, which may occur before system specific QRM [quality risk management] activities”2. The initial risk defined at the process level would be inherited across process steps, process sub-steps, associated user requirements, and functional requirements.
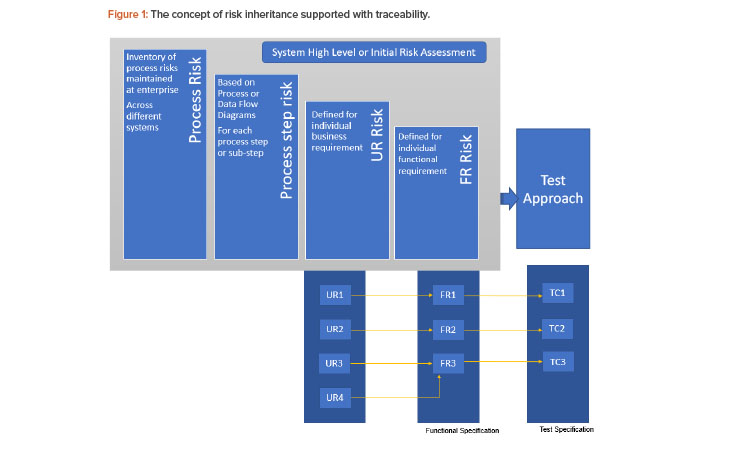
The need for the number of levels of hierarchy can be based on system and process complexity. For example, a user requirement can also be generic and linked to multiple user requirements with more specific details or directly linked to multiple detailed functional requirements. Having a baseline risk would help stakeholders only downgrade it at a subsidiary level, when necessary, without having to start the risk assessment process from scratch for every requirement. Figure 1 demonstrates the concept of risk inheritance supported with traceability.
Inherited risks vs. final risks
Inherited risks give us a good baseline risk. For any low-risk process steps or substeps, the risk assessment process may end at that level. The final risk score may still need to reassess for individual requirements for some cases.
Downgrade from inherited risks
Risks at any level classified as high may need to be revaluated at a subsidiary level to see if any subitems have a lesser risk than the overarching entity. Downgrades may be necessary in such cases and a simple principle that the substep or a subsidiary entity cannot have risk higher that the overarching process area risk can be used.
Upgrade from inherited risks
Upgrade from inherited GxP risk should seldom be used. It is important here to note that although additional flexibility is being introduced with GxP risks, other factors (including business risks and other compliance and legal considerations) should not be overlooked. Upgrade for the overall risk for the entity based on these factors may still be necessary to have a holistic control strategy.
Risk definition and inheritance process
A simple example to examine the process for risk definition and inheritance is given here and shown in Figure 2. A GxP process area can be at a high-level process (manufacturing execution, quality and batch management, etc.) or represent data like the material master.
To ensure end-to-end traceability, the traceability marker starts with a unique numbering system from each process step (simplified as 1, 2, 3, etc.). This includes a simplified model for creation/input, processing/execution, review, controls, and closure/output for a GxP master data.
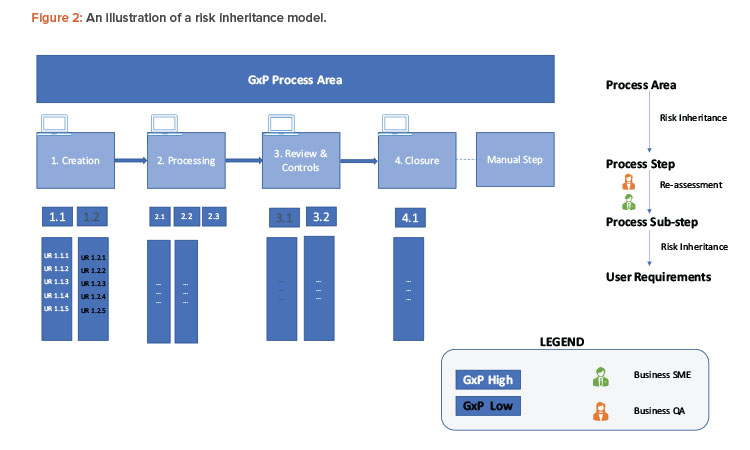
The numbering marker is inherited by all respective downstream objects like process substep (1.1, 1.2, 2.1, 2.2, etc.), user requirements (UR 1.1.1, UR 1.2.1, etc.), functional requirements (FR 1.1.1.a, FR 1.1.1.b, etc.), and test cases (TC 1.1.1.a, etc.). For example, if a process step is classified as GxP high, all of its linked objects will inherit the same GxP classification.
Inherited risks priority classification moves as is from process area to process step and is examined for reclassification at the process substep level. For example, substep 1.1 continues to have GxP high risk, but 1.2 is reclassified as GxP low risk. Any linked user requirements within substeps 1.2 are also classified as GxP low and no further GxP assessment is required at the item level. For substep 1.1, further assessments happen and require the GxP risk to downgrade to low.
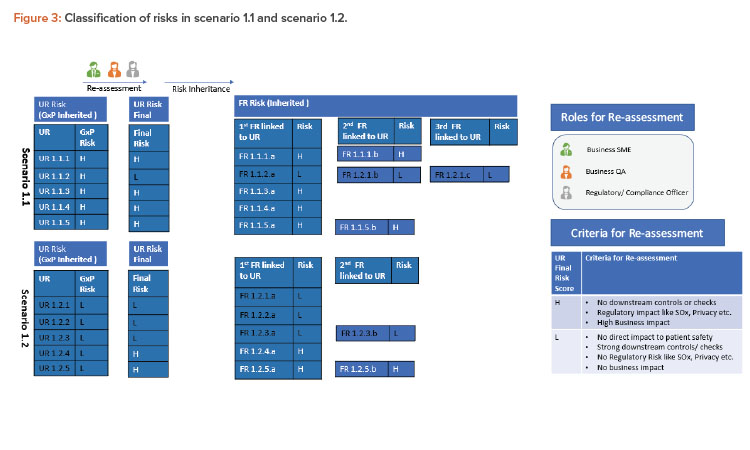
Scenario 1.1
To illustrate this further, substep 1.1 (GxP high) is considered as scenario 1.1 in Figure 3. Most of the linked requirements within this step continue to be high risk and only one user requirement (UR 1.1.2) is re-assessed as low risk. The primary consideration was that there is low GxP risk for this requirement, and secondary considerations (regulatory, downstream controls, overall business risk) were also considered by key stakeholders to conclude that the final risk for this requirement is low indeed.
Scenario 1.2
Similarly, substep 1.2 (GxP low) is considered as scenario 1.2 in the Figure 3. Here, all requirements continue as low risk with GxP. Two of these linked requirements (UR 1.2.4 and UR 1.2.5) are classified as high risk considering other factors (e.g., UR 1.2.4 was considered high due to high business risk and UR 1.2.5 due to privacy risk). The final user requirement risks are then considered to be inherited to functional requirements.
OPTIMIZING TEST SCRIPTING RIGOR
Once appropriate risks are defined and other assurance activities are in place, any items with residual risks need to be tested. The shift in focus is from providing detailed documentation regarding completion of a testing phase to improving defect detection. One of the key aspects introduced with the FDA draft computer software assurance approach and GAMP® 5 Second Edition is the use of a different unscripted testing approach for low GxP risk functionalities1, 2.
To derive the final test rigor, the functional requirement (inherited risk) is combined with the IT risk for that functional requirement as defined by the IT subject matter expert (or a group of IT experts with functional, technical, domain architecture knowledge, etc.). This is illustrated in Figure 4. Again, a critical thinking approach could be used to define IT risks. Risk is considered high for custom developments, new technologies, or systems provided by new suppliers with improvement areas during supplier evaluation. Only scenario 1.1 from the previous risk section is considered in the following description (see Figure 4).
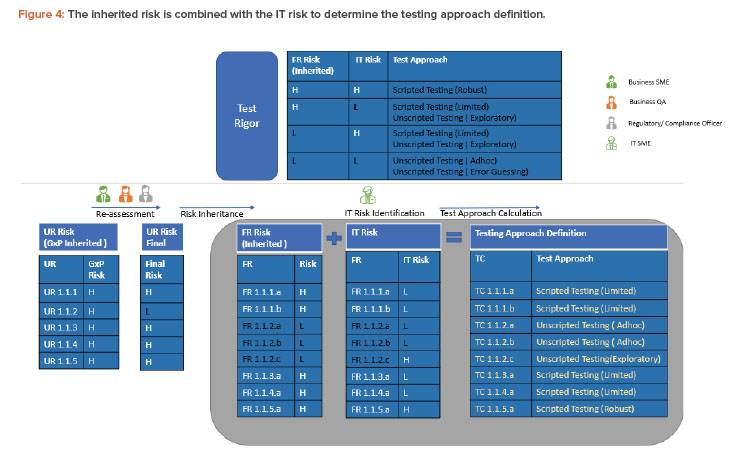
It is very clear that only TC 1.1.5.a would now require a robust scripted testing that is optimized. This is compared to the legacy approach where each of eight GxP impacted functionalities would have been tested with a robust scripting approach.
The test approach outcome here is primarily used to define the functional (operational qualification) or integration testing to be done by an IT team. The requirement for user acceptance testing (UAT, or performance qualification) is based on the process step risk. The UAT is driven by a process step risk with a simplistic approach of robust scripted testing for GxP high and scripted testing (limited) or unscripted (exploratory) testing for GxP low.
It may not always be possible to define risks at a particular level for UAT because they tend to run end-to-end scenarios covering multiple process steps. Most importantly, critical thinking should also be used to define UAT scripting approach based on parameters like experience of planned business testers, novelty of the system, etc.
A key risk to highlight here is the temptation to downgrade a custom development test approach to exploratory or ad hoc testing. This should be taken up only in a pilot and phased manner as irrespective of the GxP risk criticality, custom development brings in new exposures and effort should be made to use limited scripted testing.
This would be useful during the project phase and the operations/maintenance phase as custom developments need to be verified with different scenarios. Additionally, this is problematic when maintenance support is provided by a different vendor or team than those involved in system development phase. Critical thinking approach can be applied and simple custom developments (e.g., simple reports) can use leaner testing approaches.
AUTOMATED AND PRAGMATIC TEST EXECUTION APPROACH
Automated test execution is the best option to minimize manual overhead and is suitable for simple and integrated software suites that require multiple iterations. There is still some time before industry moves to complete automation, and where that’s not possible, pragmatic approaches to manual test execution can be applied to achieve greater efficiency.
GxP good documentation practices have heavily relied on robust test scripting as a basis for test execution with verbatim response to each test script step. A shift to a simpler scripting level approach throws new challenges into execution expectation. In the absence of a script expectation, the source of truth would move back directly to requirements and the level of detail at which they are written.
For example, a GxP low risk failure error message in the system should be exactly as aligned by business representatives, and hard coding in script would have easily detected deviations, if any were present. The error message would now need to be checked with requirement and specification documents, and this can be facilitated by taking the respective requirement ID or specification ID as an input parameter during execution.
A decrease in scripting robustness would also require an increase in robustness for writing unique testable user and functional requirement statements, supported by solid functional and quality reviews to ensure the business expectations are clearly enumerated and met. Additionally, the focus would be back on a more robust and extended UAT phase with additional time after the UAT phase for fixing any new defects.
Automated test execution is the best option to minimize manual overhead and is suitable for simple and integrated software suites that require multiple iterations. There is still some time before industry moves to complete automation, and where that’s not possible, pragmatic approaches to manual test execution can be applied to achieve greater efficiency.
Another risk associated with the usage of unscripted testing is the availability of experienced functional users for testing. Due to the volume of activity during the testing phase, junior resources are often onboarded in testing phases to follow the scripted test manual and execute tests. Care must be taken to only use experienced testers who are part of the functional design process, to achieve the expected benefits from unscripted testing approaches. In cases where this is not possible, robust training should be considered before assigning unexperienced users for unscripted testing.
It is also paramount to use pragmatism to adjust the expectation for capturing test execution. A few key areas of improvement include the following.
Recording Detailed Observed Results
Traditionally, GxP good documentation practices required actual observed test results for each step to be written in detail as per the expected evidence. A more pragmatic approach is described in GAMP® 5 Second Edition where “a simple ‘pass’” can be recorded when the system response matches the expected result and detailed statements are provided only for “failed” steps, adding additional notes for root cause analysis2.
Recording Observed Results Contemporaneously
Good GxP and data integrity practices expect results to be recorded contemporaneously. This is sometimes abused with the expectation of recording the observed result in the same minute of taking the screenshot. Although digitization of testing has helped capture the exact time stamp of the execution, some leniency in line with a paper execution could still be provided.
In a traditional paper-based wet ink execution, a tester might execute testing in the morning, take screenshots, and write the observed results sometime later the same day. A pragmatic approach can be adapted in cases where the tester missed attaching a screenshot or did not take screenshots out of the testing tool at the exact minute and second. This would also allow the tester to provide delayed additional screenshots without a full re-execution on the same day.
Screenshot Expectation
Legacy testing has heavily focused on print screen attachments as the basis for objective evidence provided for quality review. As per GAMP® 5 Second Edition, “prolific screen shots do not add value and are unnecessary” and “test evidence is only collected for proving steps that are not inherently covered by evidence from another step”2.
INTEGRATING EFFORTS WITH CYBERSECURITY AND OTHER REGULATORY UNITS
With the Pharma 4.0™ focus on having a smart factory and extensive usage of innovative tools, cloud, and the Internet of Things, there are evolving cybersecurity challenges. As per GAMP® 5 Second Edition, for GxP computerized systems, “it is usually the case that risk from cybersecurity threats directly correlate to the data integrity considerations of GxP risk”2. There is considerable equivalence of requirements between cybersecurity controls like ISO 270018, NIST SP 800-539 (with embedded privacy controls in Revision 5), and pharma regulations like 21 CFR Part 1110 and Annex 1111. This equivalence is expected because they are derivatives from the ITIL framework.
This brings good potential for synergy between an information security management unit (responsible for the confidentiality, integrity, and availability of systems) and a quality assurance compliance unit across multiple scenarios. This includes audit trails, security, data exchange, infrastructure, etc., as enumerated in a recent iSpeak blog12. The Validation 4.0 model must integrate cybersecurity and leverage activities of the information security organization to reduce redundant checks and achieve faster compliance. Combining risk assessment efforts with other compliance units achieves a holistic risk baseline for a comprehensive assurance effort required.
CONTINUOUS CONTROL AND COGNITIVE COMPLIANCE
The usage of tools and artifacts supports creation of real-time traceability and test reports, which are readily available for self-assessments. According to a 2021 Pharmaceutical Engineering® article, “instead of relying on difficult to maintain silos of documentation, we look toward digital artifacts managed with appropriate tools that can instantaneously provide reporting and notifications on the state of control”4. Carefully configured validation checks can highlight discrepancies in the updating of linked objects. For example, if a functional specification is updated without updating the l inked design specification, it will be flagged as an issue item for consideration.
Key quality metrics can be defined and compliance dashboards can be created, with the option of filtering and reporting. Parameters can be set based on the number of red flags highlighted, and a group of objects or a project may be zeroed down as a potential audit candidate that will feed into audit plans. Continuous process verification (CPV) is considered key for Validation 4.04. The same principle should be applicable for CSV to ensure key validation documents and that the metadata and traceability is in a continuous state of control.
Other Pharma 4.0™ priorities like the usage of artificial intelligence (AI) can also be used to gain insights that were not possible before5. Although document content reviews may still be done manually, there is a lot of potential with advent of AI. Simple good documentation practice checks (e.g., usage of templates, filling of all subsections, expected metadata values in some fields, blank spaces, date format, and other specific checks based on document types) can be easily automated. This will help quality reviewers to validate in conjunction with manual review.
With ChatGPT having revolutionized all industry areas, a similar AI tool can also be used in validation context. Typical inputs for this tool can be a high-level project documentation with project context and answers to some key questions with the output as a project documentation list. This should set the context for the minimum required deliverables for a lean validation approach with any additional documents only added in scope with appropriate justification.
HOW THE SEVEN PILLARS SUPPORT THE PHARMA 4.0™ OPERATING MODEL
It is important to note how these pillars support the key themes of the Pharma 4.0™ operating model.
Holistic Control Strategy
As noted in a previous Pharmaceutical Engineering® article, “the validation strategy must be part of the holistic control strategy, and stakeholders must use critical thinking to ensure lean and robust risk assessment”3. Furthermore, this article also notes that holistic control strategy enables data-based decision on all aspects of the pharmaceutical product life cycle. The same holistic approach can be extrapolated to the computerized system life cycle with data derived from software tools as the key drivers for real-time dashboards and continuous audits.
Another aspect for holistic control strategy and integrating efforts is leveraging efforts between different compliance groups within the pharma organization—GxP compliance, cybersecurity managers, and other regulatory compliance officers to avoid redundancy of checks and duplication of efforts.
Data Integrity by Design
Process and data flows form a key part of ensuring data integrity by design because clear understanding of the flow can help to detect any integrity flaws and mitigate them. Prospective data integrity is a key feature of Pharma 4.0™13, and usage of software tools and digital documentation ensures data integrity for validation documentation is foolproof and detailed. Tool-based digital documentation data is legible, attributable from the time it’s recorded, contemporaneously recorded with real-time version history, stored in the original format, and contains validation checks for accuracy.
Digital Maturity
Moving to predictive capability and adaptability can be achieved through real-time dashboards. In terms of computer system validation tools, maturity needs to move from Level 2 and Level 3 to Level 4. A shift from Pharma 3.0 (until Level 2) to Pharma 4.0 (Level 3 onward) is a business imperative for companies moving to niche products or advanced therapy medicinal product needs13. Some pharmaceutical companies are already at Level 3 to some extent, but a complete transition needs to happen.
Moving to the new approach would require validation managers to develop the skillset of techno-functional business analysis in challenging the risk assessments more confidently. This would allow them to set the baseline for a risk-based testing effort and to become champions to facilitate the critical thinking approach.
The critical thinking approach in this article has only been discussed in a limited context, but the use of a rational and a well-reasoned approach adds value across all aspects of CSA6. Yet another key enabler could be the close involvement of reviewers in training and setting the right expectation before the start of the activity. This would ensure document definitions are correct from the beginning, with iterative reviews happening to avoid rework. There are other enablers in terms of management support and regulatory feedback, and publication of the final CSA guidance will hopefully provide a definitive conclusion to this conundrum.
CONCLUSION
CSV has been interpreted and used in myriad ways in the pharmaceutical industry, with stringent organization-specific conventions and practices sometimes exceeding regulatory mandates. This has burdened delivery teams with unrealistic checklist-based expectations. GAMP® 5 Second Edition made a key contribution in this regard by highlighting practices that are overhead tasks and not truly required to fulfill regulatory expectation.2
One of the main objectives of Industry 4.0 is to build a lean digital core, and a primary value of the Validation 4.0 approach would enable a lean validation approach, supporting establishment of this lean digital core. The key pillars highlighted support early identification of risks. This enables ringfencing of high-risk individual items and the required assurance activities without impacting full related functionality.
Furthermore, the industry’s focus is shifting to extensive defect detection without producing piles of documentation to demonstrate compliance. Some challenges will remain, especially with respect to the acceptance and adoption of the new approach, the organizational change management, and the cost of tools and integration. References


