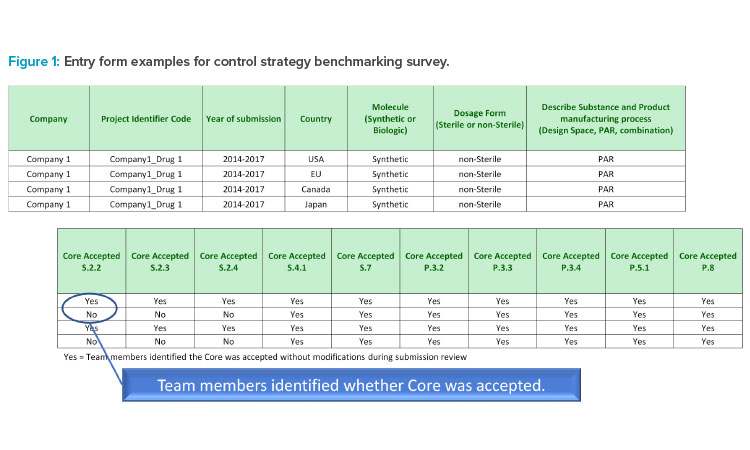
The data shows wide variation in acceptance rates and overall low acceptance rate of documents that industry believes meet ICH control strategy requirements based on their acceptability in at least one ICH region. This is concerning in light of the well-established nature of these regulators within ICH. When considering new ICH regulators and observers, it is reasonable to assume the overall acceptance rate may drop significantly. Industry has an important harmonization strategy to develop and use a single set of control strategy documents, but regional and local preferences drive a plethora of additional commitments.
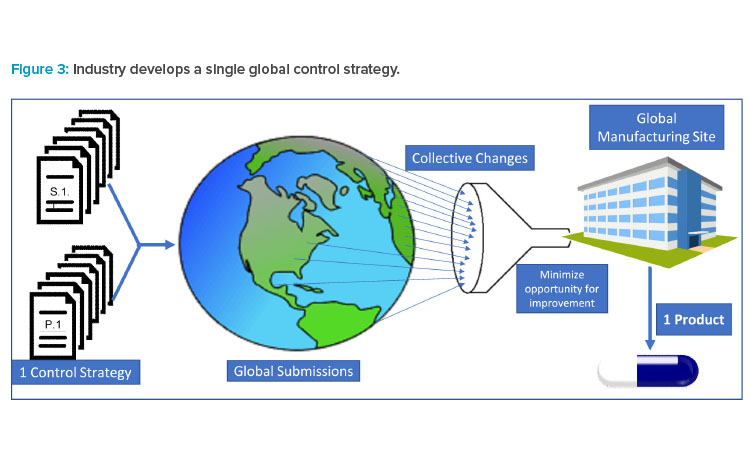
Pharmaceutical products are typically globally manufactured and released for all marketed countries, not a single or group of countries. As a result (and as illustrated in Figure 3), industry must collate all requested modifications and requirements to create a single set of requirements for “one-product manufacturing.” Industry must then accommodate regulatory requests by amending CMC controls or segregating materials for supply in a specific market.
Thus, differing accepted test methods and specifications become barriers to innovation and continual improvement for all global products and patients.
Control Strategy Divergence and Patient Impact
The lack of harmonization of control strategies delays access of new medicines to global patients. Applicants must stagger global submission plans for new medicines to allow time to answer questions from global quality regulators. Although applicants submit a single core control strategy to supply the global markets, the same science, justification, and data sets are often interpreted differently by health authorities. This difference in interpretation of control strategy suitability results in the high degree of variability in the volume of questions from global quality reviewers on the same set of documents. Frequently, an applicant will not only experience a large variation in the number of questions, but will also encounter multiple rounds of questions from a given regulator on a particular topic. Additionally, some health authorities have limited windows for an applicant to respond to questions.
As a result, subject matter experts with specific knowledge of the process, testing, and specifications must prioritize preparation of responses over other R&D efforts and spend many hours to provide additional justification for the control strategy that was submitted and/or implement control strategy changes. The time spent on these efforts limits the ability of a company to submit global applications and inhibits further development and expansion of supply that would allow global patients to have access to potentially life-saving medicines. Among the companies that participated in the control strategy survey, many observed a drastic difference in the number of questions received on the same data between agencies (for example, 19 from one agency and 184 from another). The difference in questions is all too common and reflects notable differences assessing suitability of the control strategy.
Country-specific control strategy requirements can also potentially lead to a drug shortage if material made and released for one region is unsuitable for a different jurisdiction. Pharmaceutical products are typically manufactured for global release, not for a single or group of countries. Local constraints on control strategy, such as tightened manufacturing ranges and/or specifications based on a limited number of manufactured batches, are especially costly. Alternatively, a comprehensive science- and risk-based approach is strongly favored. Although additional control strategy requirements by any given country can be accommodated, the combined requirements of over 100 countries can add significant manufacturing and supply barriers.
The lack of a single control strategy for all countries will lead to needless supply chain complexity and can have profound impact on supply of critical medicines to patients. Although companies may manufacture to the most stringent control (parameter range or specification limit) in the case of failure to meet the tighter controls, country-specific release may be applied. However, country-specific release is inherently a complex process because it is intended to be used by exception and could delay the release of product for distribution. For products with supply constraints due to manufacturing capacity, complex manufacturing process, or unforeseen supply chain disruptions, this can lead to potential stock-outs.
Sponsor X reported a case in which it was requested that an impurity specification limit, based on available batch data, be tightened, even though the process had been demonstrated to tolerate a higher limit consistent with safety considerations. In this situation, a tighter impurity limit for the intermediate would have led to a delay in availability of the new medicine in this country because the product for the launch was made with intermediate that did not meet the tightened specification. The company’s rationale for keeping the originally proposed specification was accepted, but often sponsors are forced to accept a lower limit.
ICH Considerations
Expansion of new ICH members and observers is expected to result in continued escalation of divergence and increased obstacles to realizing globally harmonized control strategies. New ICH members have the challenges of adopting ICH guidelines while building internal capability to use them properly, which creates at least temporary divergence as the health authority transitions to the ICH-enabled future state.
At the time of marketing applications, when there is limited experience and data, constraints imposed by global regulators on licensed control strategies are inconsistent with ICH and limit innovative changes after approval. One example of such limitations is how companies describe their product manufacturing controls in the marketing authorization application. Using process control terminologies—such as proven acceptable range (PAR), normal operating range (NOR), or design space (DSp)—resulted in varying interpretations of ICH guidance and led companies to abandon strict adherence to terminology to instead focus on basic scientific principles and a robust, well-controlled process. When scientific principles are applied, some health authorities insist on applying these categories in assessing applications within their jurisdiction.,
Divergent interpretation and implementation of ICH guidelines among regulators is therefore a growing problem for industry. Applicants are typically left with unnecessarily constrained control strategies, which can limit shelf life, reduce process capability, and restrict changes that could otherwise be implemented through a pharmaceutical quality system. Applicants are frequently left with no option but to accept a country-specific control strategy requirement rather than risk product nonapproval or delays to approval. Examples related to selection and justifications of drug substance starting mate-rials clearly demonstrate the negative impact of such divergence.
As an example, Sponsor A proposed two crystalline products as starting materials that were justified by ICH Q11. Regions/Markets A, B, and Q did not query the sponsor’s starting material strategy; Market E was not satisfied with the proposal and requested more of the synthetic steps to be put under GMP control, stating that the proposed regulatory starting materials do not meet ICH criteria, given short synthetic routes from each starting material to the drug substance. The sponsor acquiesced to Market E, defining submissions with starting materials consistent with Market E’s preferences, creating a divergence of the control strategy that was approved in other markets. Additional drug substance process performance qualification (PPQ) requirements and other control strategy adjustments can lead to delayed approvals and delayed availability of new medicines for patients.
Similar divergence was observed in a presubmission advisory meeting. Sponsor B proposed starting materials of a synthetic drug substance consistent with ICH Q11 and sought agencies’ advice. Agency X agreed with the proposed starting materials. Agency Y did not agree with the proposal and required additional steps from the syntheses of the starting materials to be under GMP control. Agency Y’s view was that there was no way to track changes to the starting material processes in the absence of GMP control, which is inconsistent with the intent of ICH Q11. Control strategy changes were made and additional PPQ on drug substance was conducted to satisfy Agency Y. A separate marketing application with revised starting materials was submitted for Market Y, though acceptance of the original starting materials would have provided for earlier submission and approval.
Although not directly part of the survey, recent regulatory response to submissions for COVID-19 vaccines presents an interesting and positive example. Companies initially sought Emergency Use Authorization (EUA) rather than normal submission processes, and supplies were being provided from a clinical manufacturing process. The ability to gain broad approval with a single control strategy has allowed rapid distribution of the vaccines throughout the world. Had the EUA approval process been slowed by protracted control strategy queries, it is possible that access to vaccine would be limited, even now. Increasing efforts toward convergence, a collaborative review, e.g., Orbis, will improve harmonization and lessen divergence. Historically, industry CMC organizations have not held up submissions due to numerous changes to the control strategy caused by divergent interpretations of ICH guidance, but rather as more innovative technology is used to manufacture pharmaceutical products, it will become more challenging for industry to be able to accommodate local needs without resulting in delays.
Continual Improvement and Impact on Postapproval Changes
After a product has been approved by the regulatory agencies, it is standard to make improvements in the pharmaceutical manufacturing process to increase production scale, or to implement technological advancements such as real-time testing, or to modify control strategies.
Continual monitoring efforts are in line with ICH Q8, Q9, and Q10 guidance: ICH’s Questions & Answers for ICH Q8, Q9 and Q10 states:
Continual monitoring (e.g., via Continuous Process Verification) can further demonstrate the actual level of assurance of process consistency and provide the basis for continual improvement of the product. Quality Risk Management methodologies of ICH Q9 can be applied throughout the product life cycle to maintain a state of process control.
A holistic approach to quality improvements as described in the ICH guidance is the desired state for a robust quality improvement. The FDA and EMA further describe the principles of continuous process verification and how companies may take advantage of new advancements when applying enhanced process understanding coupled with risk management tools and a pharmaceutical quality system. Application of continual improvement in the current regulatory environment is a challenge, made more so when a product has customized controls for multiple markets.,
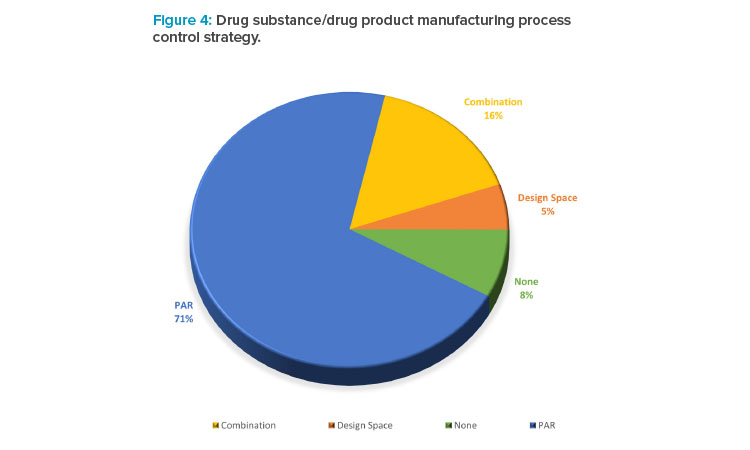
In the collected data set, companies were asked to describe their drug substance and drug product manufacturing process (Figure 4). The data illustrated that the term of design space is not used as frequently as PAR, despite most companies routinely undertaking some degree of enhanced development and studying interactions between process variables and product quality in developing the control strategy.