Leveraging GAMP® 5 Second Edition for Medical Devices

This article provides a brief introduction into the standards and regulations for medical devices. It compares the ISPE GAMP® 5 Guide: A Risk-Based Approach to Compliant GxP Computerized Systems (Second Edition) and applicable ISPE GAMP Good Practice Guides against the relevant regulations and standards for the development of software for medical devices and demonstrates GAMP® 5 Second Edition’s applicability.1
The standards discussed in this article are 21 CFR Part 8202 and ISO 13485:20163 for the quality management system, IEC 623044 and IEC 82304-15 for the software development life cycle, and ISO 149716 for the application of risk management to medical device software.
The standards for the design and development of medical devices and GAMP® 5 have different focuses. To provide the necessary understanding, the design process for medical devices, the risk management process applied to medical devices, and the software development process for medical device software are first introduced, and the system life cycle is applied to the GxP-critical systems of GAMP® 5 Second Edition for comparison.
Opportunities for leveraging GAMP® 5 Second Edition for medical devices and vice versa are also identified in this article. Steps for optimization will be proposed and can be applied in scenarios where the advantages of GAMP® 5 Second Edition are leading, e.g., inclusion of an IT system used as the backend of a complex medical device, or for inclusion of a mobile device managed according to the GAMP® Good Practice Guide: A Risk-Based Approach to Regulated Mobile Applications.7
Validation of Supporting Software and Computerized Systems
As in pharmaceutical manufacturing, where computerized systems “used as part of a GMP regulated activities”8 must be validated, quality management systems for medical devices require validation of systems used in production, quality management, or service provision.
This list can also include systems that are used in the design and development of medical devices, as demonstration of conformity of the designed product includes a demonstration that the systems, instruments, and software used to support product design and development are fit for their intended use. This is comparable to pharmaceutical industries, where GAMP® 5 Second Edition is not only applied to systems used in manufacturing processes, but also to systems in preclinical research and drug development.
Although it was originally created for the pharmaceutical industry, GAMP® 5 Second Edition may be applied for systems supporting the life cycle of a medical device. Usage of GAMP® 5 Second Edition for this purpose is widely practiced and accepted in medical device industries.
The first edition of GAMP® 5 excluded “software embedded within medical devices,”9 but the scope of GAMP® 5 Second Edition now explicitly includes “Medical Device Regulations (where applicable and appropriate, e.g., for systems used as part of production or the quality system, and for some examples of Software as a Medical Device [SaMD]).”1
This broadening of the GAMP® 5 Second Edition scope was also already anticipated by the publication of GAMP® Good Practice Guide: A Risk-Based Approach to Regulated Mobile Applications in 2014.7 With a detailed comparison of the standards explained in the following sections, we can outline the steps to harmonize a company’s software and IT activities.
Alternatively, ISO/TR 80002-210 may be applied for validation of systems supporting the medical device life cycle. This is not a harmonized standard, but rather a technical report that acts as a guideline like GAMP® 5 Second Edition. In terms of content, it is very close to the US AAMI TIR36,11 published in 2007, which is a Recognized Consensus Standard used as a methodological basis by the FDA for the medical device industry.
Medical Devices: Standards and Regulations
Medical devices are devices used according to the intention of the manufacturer for the diagnosis, treatment, or monitoring of patients. To minimize risk for the patient, medical devices are controlled by regulations and standards.
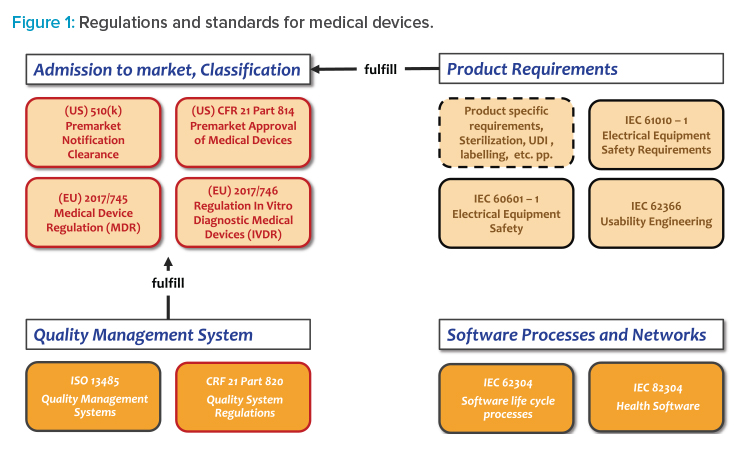
Regulations and standards include general regulations for authorization of medical devices to the market, implementation of a quality and risk management system, medical device product standards, and more. If the medical device includes software, process standards for development and maintenance of software are also included (see Figure 1).
Understanding Life Cycles
It is essential to realize that GAMP® 5 Second Edition and the medical devices standards deal with different objects and processes. Consequently, a simple ad hoc mapping of activities, artifacts, and documents must be avoided. On the other hand, both GAMP® 5 Second Edition and the medical devices standards must manage and control software development and implementation. It is necessary to compare the different regulatory landscapes to understand how each approach to software development and maintenance disciplines varies.
The following discussion displays for each examined medical device process the controlled objects and scope of the life cycle progression, providing a structured approach for comparison with validation life-cycle of GAMP® 5 Second Edition, where the controlled object is the computerized system, and the scope of the life cycle extends from the initial concept through implementation and operation to retirement or replacement of the system.
Design Control for a Medical Device
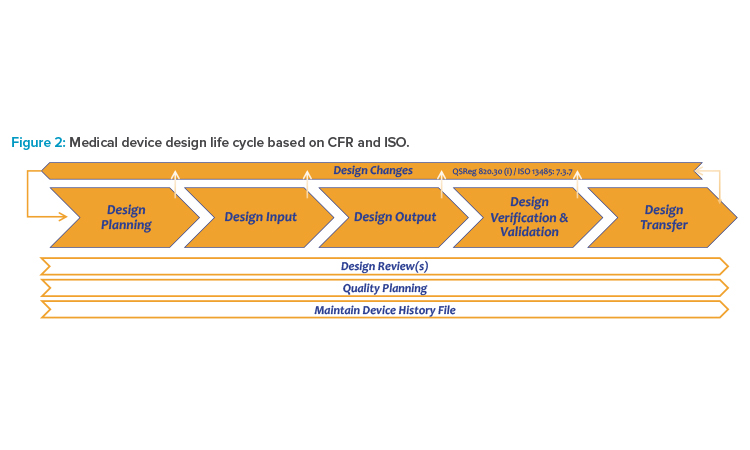
The quality management system for medical devices, according to ISO 13485:20163 and 21 CFR Part 820,2 includes the process for design control of a medical device from initial planning to transfer to production (see Figure 2).
- The controlled object in design control is the medical device as dedicated for the market according to its classification, e.g., according to Appendix IX of the EU Medical Device Regulation.12 The medical device can include mechanical, electrical, and software components.
- Software specifications are defined as part of the design input. The fully developed software package ready for installation into the medical device is part of the design output. Typically, a medical device includes software, mechanical or electrical components, and accompanying documentation.
- Design control applies if a product is intended to be a medical device and is classified respectively as a Class I, IIa, IIb, or III medical device. Manufacturing, packaging, labeling, storage, installation, and servicing of medical devices are not part of design control.
- The scope of the design control process is the design of a medical device, starting with its initial planning and classification and ending with its transfer to production. The approval of a medical device for the market is handled separately but relies on the correct application of design control.
- The correct design of a medical device is controlled by its related design history file, which covers all phases of the design control process, including verification and validation. In the case of possible design changes, the design control process is triggered again.
Software Development Life Cycle: IEC 62304
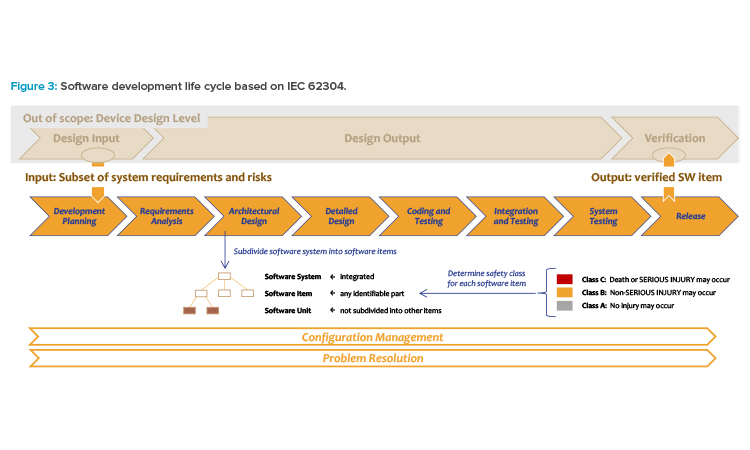
The IEC 62304 standard covers the controlled development of software components (software implementation) for medical devices from planning to release, as well as subsequent management of changes or software errors (see Figure 3).4 Software implementation fills the gap between design input and design output in design control.
- The controlled object in the software development life cycle is the software item, i.e., one node or component of a software system. The software items are defined in the related software architecture. If the controlled software item is the root element, the process covers the complete software system.
- The software development life cycle is not an independent life cycle. It inherits the software requirements from design input. The fully developed software package becomes part of the design output.
- The software items are assessed according to their safety classification (see safety classes as defined in IEC 62304). Each software item of the software system can be assessed separately. The safety classification is different from the classification of the final medical device.
- Scope of the software development life cycle begins with documentation planning and requirement analysis and ends with release of the integrated and tested software item for installation into the medical device. In case of changes or correction of errors, the software development life cycle is triggered again.
- The documentation of the software development according to initial documentation planning becomes part of the design history file of the medical device.
IEC 62304 Standard Processes
The IEC 62304 standard focuses on the software development process and defines the typical activities of the development life cycle such as planning, requirements analysis, design, implementation, verification and testing, and release.4 (See the previous description of the software development life cycle.)
Overall, the standard describes process and documentation requirements for each phase of the software development life cycle and covers five processes:
- The software development life cycle process
- The software maintenance process
- The software risk management process (includes a reference to ISO 14971)
- The software configuration management process
- The software troubleshooting process
The IEC 62304 standard does not stipulate a specific process model for the software development life cycle (like waterfall, V model, agile, or scrum). Instead, it contains requirements for specific activities and development disciplines and its documentation.4 These activities represent minimum requirements for contemporary software development.
A relatively large section of IEC 62304 standard is dedicated to maintenance and troubleshooting processes. Medical device software issues are still a major driver in device recalls.
The IEC 62304 standard is based on the international standard for software life cycle processes, ISO/IEC 12207,13 so it should not be difficult to harmonize with the GAMP® 5 Second Edition approach for software documentation and to use common life cycle templates to cover both fields.
The IEC 62304 software risk management process requires that criticality is always evaluated, i.e., the extent to which the software could be the cause of a hazardous situation for the patient.4 This evaluation must be documented in the risk management cycles.
Risk control measures derived from the evaluation must be implemented, verified, and documented in such a way that full traceability between hazard and product requirements that mitigate the hazard is kept throughout the full product life cycle. In this sense, a risk in scope of the risk management is also always a design risk. Other risks, e.g., project risks, are managed separately. These risks are not in the scope of the IEC 62304 or ISO 14791 standards.
Safety Classes as Defined in IEC 62304
To minimize the effort and expense involved in documentation, the IEC 62304 standard defines so-called safety classes (see Figure 3 ) from Class A to Class C, reflecting increasing severity of possible harm to the patient.4 The higher the safety class, the more completely the aforementioned specifications of the standard must be implemented. For example, IEC 62304 requires only the software specifications and software approval for Class A software items; even testing is not required except testing of the fully integrated system [4].
The safety classes must not be confused with the medical device classification previously described, e.g., Class I, IIa, IIb, and III, based on the level of control necessary to assure safety and effectiveness, ranging from low to high risk. The medical device classification is applied to the product, whereas safety classes are applied to one clearly identified software component: the software item (see Figure 3).
It is the responsibility of the manufacturer to establish the definition and granularity of the software system and to document it in the software architecture.
The standard uses three terms to describe the breakdown of a “software system” defined as the fully integrated software. The software system can be a subsystem of the medical device or a stand-alone medical device.
A software system consists of one or more software components, called “software items,” and each software item may also consist of one or more software items. “Software units” are software items that cannot be further broken down. It is the responsibility of the manufacturer to establish the definition and granularity of the software system and to document it in the software architecture.
As previously discussed, the standard is applied not only to the integrated software system but to each software item with assigned safety class, i.e., each software item must be able to successfully pass the software development life cycle process. This introduces a high level of segregation and control of dependencies between software items.
Typically, if software items that do not have a parent-child relation must interact, the software architecture will define an internal interface. Segregation of software items is a safeguard to ensure that high-risk software items are not accidentally impacted by lower risk software items.
Probability or detectability play no role in determination of the safety class; it deals only with consequences with respect to the patient. Also, data integrity and product quality, known as criteria for risk determination for GxP-critical systems, only play a role if they have consequences for the patient.
Health Software Product Life Cycle: IEC 82304
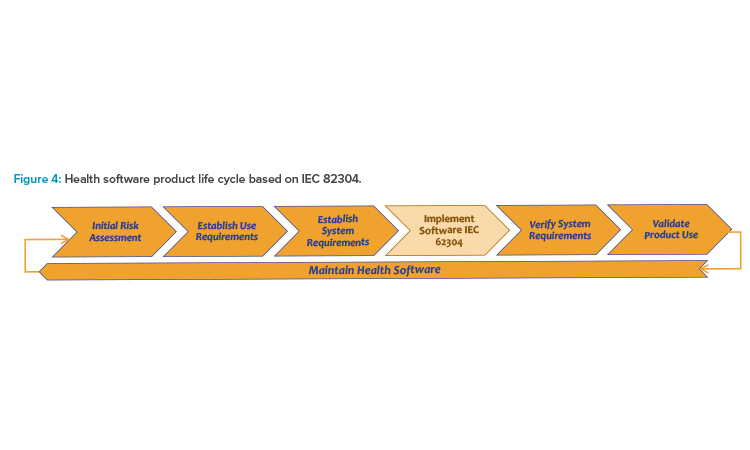
The IEC 82304 standard extends the software development life cycle previously described as applied to a software-only product (see Figure 4).5 It is an interpretation of the design control process for software as medical device and a completion of the development life cycle for health software products that are not classified as medical devices. The following description focuses on medical devices.
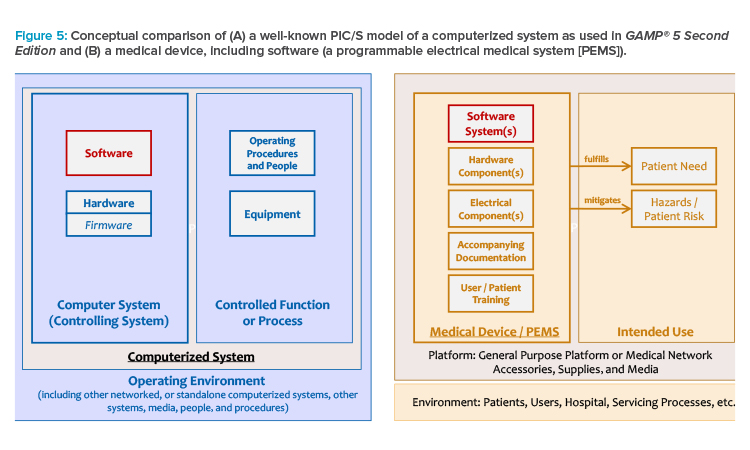
A medical device intended to fulfill a specific medical purpose can be considered conceptually equivalent to a computer system intended to control a specific function or process.
- The controlled object is a health software product, which may include SaMD or other health software not classified as medical device. In the case of a medical device this means it is a fully stand-alone SaMD. Respectively, the health software product life cycle complies with the design control process for medical devices.
- For software implementation, the health software development life cycle relies on the software development life cycle according to IEC 62304.4
- If the health software product is intended as a medical device, it must be classified according to the medical device regulations.
- The scope of the health software development life cycle for the case of medical devices is the design control, starting with initial risk assessment and establishment of product requirements up to validation of the software product as basis for its release to market as medical device.
- The correct development of the software product for a medical device is controlled by the design history file. Contributions according to IEC 823045 are the software requirements and the final validation report. In case of possible design changes, the process is triggered again.
Computerized System vs. Medical Device
For further understanding, the controlled objects treated in the life cycle models are compared based on the Pharmaceutical Inspection Co-operation Scheme (PIC/S) model for computerized systems in regulated GxP environments (see Figure 5).
A medical device intended to fulfill a specific medical purpose can be considered conceptually equivalent to a computer system intended to control a specific function or process. The patient risk of the intended use of a medical device plays the same role as the GxP criticality according to an initial risk assessment for a computerized system.
Software is a key component in a computerized system, as well as in a PEMS. A medical device may consist only of software components (SaMD) that are installed on a general-purpose platform, like a mobile device, or are deployed with a medical network.
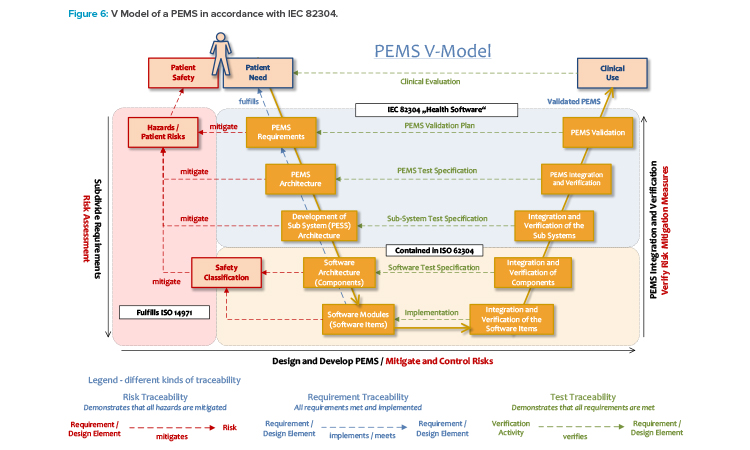
Combining IEC 62304 and IEC 82304
Requirements for software are only a subpart of the requirements for a PEMS:
Figure 6 shows a schematic diagram of a V model of PEMS development. The specifications of IEC 62304 apply only to the PEMS component level and below. Validation is defined as the “evaluation of whether a product meets the requirements for the intended purpose”14 and the “confirmation […] that the requirements for a specific intended use or a specific intended application have been met.”3
That is, validation requires a clearly defined intended use and valid requirements for use. As IEC 62304 focuses particularly on software items embedded in medical devices, requirements for software verification are formulated, but not for validation. Therefore, medical device manufacturers can rely on the standard IEC 82304 (“Health Software”), which covers the top section of the V model in Figure 6 and is also applicable to stand-alone software, or SaMD. 5
Conclusion
We have shown that GAMP® 5 Second Edition can be fully compatible with medical device needs when a few points or gaps are correctly understood and appropriately addressed. Bringing the worlds of GxP-critical IT systems and medical device software together requires limited initial effort.
As soon as these steps have been taken, the advantages of GAMP (e.g., in system operation, management of critical data, early quality involvement, and leveraging of supplier activities) are revealed for the medical device world. The GAMP world can take advantage, in particular, of medical devices teams’ expertise on software architecture and segregation, management of design risks, and software development.
An in-depth comparison of the medical device processes with GAMP® 5 Second Edition including a roadmap for harmonization will be given in an upcoming ISPE/GAMP concept paper.
Acknowledgments
The authors would like to thank the members of the Germany/Austria/Switzerland (D/A/CH) Affiliate Application of GAMP 5 in the Medical Device Field special interest group (SIG) for their continuous support and contributions.


