Making CPV a Proactive Component of Process & Product Improvement

Change often takes years longer in the pharmaceutical industry than in others. Why can we not challenge that paradigm? The auto industry, for example, successfully forced changes to its supplier base within a couple of years. By benchmarking other industries that have dealt with similar problems, we can learn from their experiences.
Our purpose in this paper is to aid pharmaceutical companies in their CPV journey by sharing the lessons we have learned during our combined 70 years of practical experience. Although the content of this paper is primarily technical, we have added comments to illustrate the merits of this discussion where appropriate. We would have liked to include case studies and discussions of other topics, but that would have required much more material than space allows.
While continued process verification (CPV) may be relatively new to the pharmaceutical industry, it is not new to most others. The automotive industry and its supplier base, for example, began to implement such programs in the 1980s. One of us wrote a book chapter on this subject in the early 1990s.1 Building on that base, the current paper focuses on three key points:
- Monitoring can mean many things, not just control-charting.
- Process control strategy should be viewed as a living, risk-based business process subject to constant review and potential revision.
- Industry should focus on the business process side of CPV, constantly improving its efficiency and effectiveness.
POINT 1
Monitoring ≠ control-charting
It is quite clear from our experiences in the pharmaceutical industry that “monitoring = control-charting” is a common mindset. We’ve seen this in a variety of ways:
- Auditors: “What is being monitored? Show me the control charts.”
- The CPV monitoring plan showing variables as either “in” or “out.”
- “We are monitoring critical quality attributes and a few critical process parameters.”
This is an inadequate way of thinking. Monitoring is not a yes-or-no proposition, but a continuum—a view of the concept that has been utilized in the automotive industry for decades.2
As an analogy, imagine going to the hospital and hearing a doctor ask a nurse to begin monitoring the patient’s blood pressure. The doctor does not mean the nurse should pull out a control chart. Rather, “monitor” indicates an appropriate level of observation, whether by cu once an hour, twice a shift, or by automatic continuous monitoring.
The US Food and Drug Administration (FDA) recognizes this distinction. Consider, for example, the agency’s guidance for process validation:
The terms attribute(s) … and parameter(s) … are not categorized with respect to criticality in this guidance. With a lifecycle approach to process validation that employs risk based decision making throughout that lifecycle, the perception of criticality as a continuum rather than a binary state is more useful. All attributes and parameters should be evaluated in terms of their roles in the process and impact on the product or in-process material, and reevaluated as new information becomes available. The degree of control over those attributes or parameters should be commensurate with their risk to the process and process output. In other words, a higher degree of control is appropriate for attributes or parameters that pose a higher risk. [emphases added]
Later in the document, the FDA states that monitoring levels should be adjusted according to performance, not just in or out of the monitoring program:
These estimates can provide the basis for establishing levels and frequency of routine sampling and monitoring for the particular product and process. Monitoring can then be adjusted to a statistically appropriate and representative level. Process variability should be periodically assessed and monitoring adjusted accordingly. [emphases added]3
| Level | Process monitoring intensity level |
|---|---|
| 1 | Recording data manually or electronically |
| 2 | Comparing data to specification limits |
| 3 | Plotting run charts |
| 4 | Plotting against pre-control or practical alert/action limits |
| 5 | Plotting control charts having statistically derived limits |
| 6 | Operating an automated controller (feedback or feedforward) |
- 1Faltin, Frederick W., and W. T. Tucker. “On-Line Quality Control for the Factory of the 1990s and Beyond.” In Statistical Process Control in Manufacturing, edited by J. Bert Keats and Douglas C. Montgomery. New York: Marcel Dekker, 1991.
- 2Kane, Victor E. Defect Prevention. New York: Marcel Dekker, 1989.
- 3US Food and Drug Administration. Guidance for Industry. “Process Validation: General Principles and Practices.” January 2011. https://www.fda.gov/downloads/drugs/guidances/ucm070336.pdf
PMILs
Process monitoring intensity levels (PMILs) should be categorized and applied in specific instances based upon risk (see example in Table A). While the pharma industry certainly uses these control methods, they aren’t usually called out and tied to risks.
PMILs are mutually exclusive, so each variable would be assigned to only one level at any point. Over time, a monitored characteristic might move from one level to another, depending on circumstances. Each variable considered would be assigned an appropriate PMIL, based upon risk level.
This is not a question of what is being monitored, but how. As risks change, the intensity level should change accordingly. We have seen numerous examples of variables—even those with very high performance levels—being control-charted even when they exhibit very high performance levels as measured by a metric such as the process performance index, or Ppk.
| Level | Variance ratio | Standard deviation ratio | Analytical method monitoring intensity level |
|---|---|---|---|
| 1 | < 10% | < 30% | Normal system suitability testing and standards testing (with acceptance limits) |
| 2 | 10% –25% | 30%–50% | Precontrol or practical alert/action limits |
| 3 | 25%–50% | 50%–70% | Control-chart system suitability testing/standards data |
| 4 | > 50% | > 70% | Investigation or remediation required |
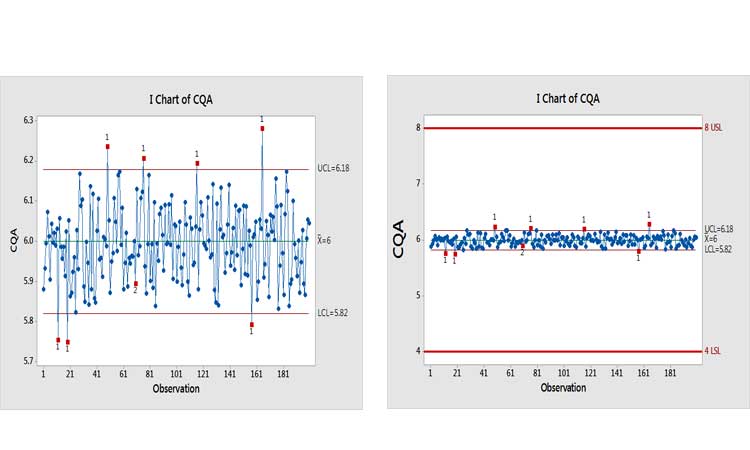
Extremely capable processes have been known in a variety of industries for decades. In cases of “excess capability,” statistical process control methods should be adapted to provide meaningful information. Control-charting was developed on the assumption that the process under study was unstable or, at best, stable but marginally capable. Manufacturers needed a way to discover changes quickly to avoid producing nonconforming product.Figure 1, for example, shows a control chart of a variable. Taken in isolation, the message conveyed seems alarming (pun intended). When placed into the context of the specifications, however, it’s apparent that this variation, whether from special or common causes, is virtually meaningless.
“Excessively capable” processes represent the opposite of this scenario. Their high Ppks demonstrate that their centerline and variability are unlikely to produce out-of-specification product. We plan to devote further attention to this issue in a subsequent paper.
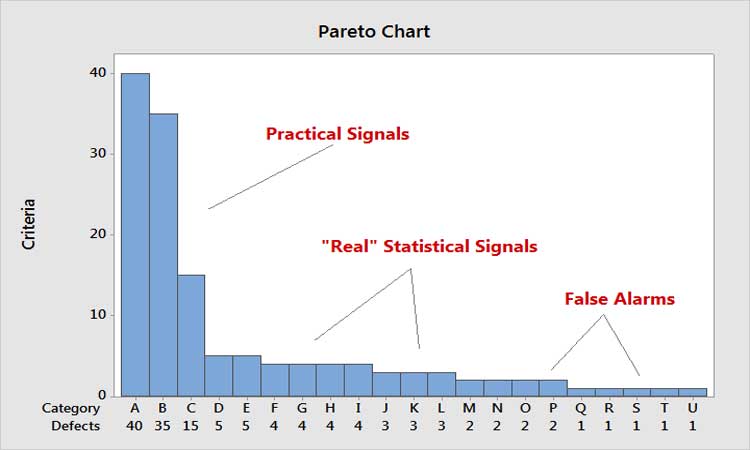
We’ve often heard the argument that specifications do not reflect what is clinically relevant. If so, the fault lies with the specifications. They should be changed, even if only internally, thereby creating a new performance (Ppk) value and risk level to drive the level of monitoring assigned. We’ve also heard the argument that every signal is an opportunity to learn, regardless of the specifications. Imagine doing an Internet search that retrieves millions of results, then sifting carefully through each one, instead of applying the Pareto principle∗ to determine the appropriate choices. In industry, over-monitoring can produce an essentially infinite number of out-of-control signals, making it impossible to devote equal energy to all of them. (We will address this and related issues below, in the section on efficiency and effectiveness of CPV as a business process.)
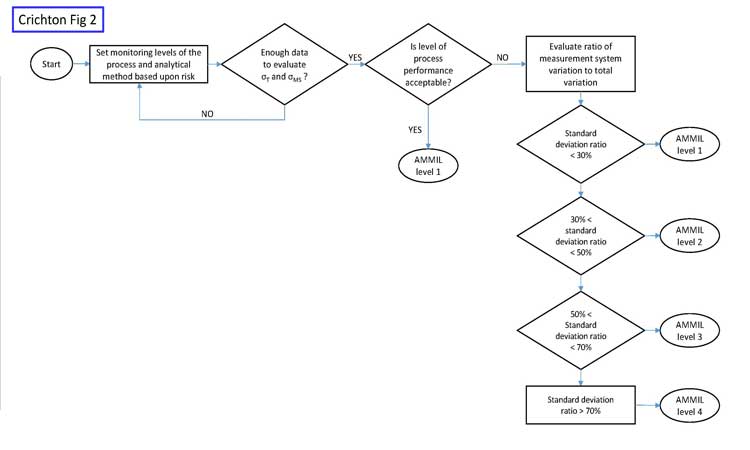
These same ideas apply to CPV monitoring of analytical methods. Risk levels should be evaluated by measuring the influence of the measurement system on release data. One way to do this is to calculate the ratio of the measurement system variability (_lowersigmaMS or _lowersigma2MS) over the total variability (_lowersigmaT or _lowersigma2T) of the release data. In Six Sigma literature, the ratio of standard deviations (_lowersigmaMS or _lowersigmaT), when expressed as a percentage (Table B), is called the “percent repeatability and reproducibility,” or simply %R&R. An analytical method monitoring intensity level (AMMIL) can then be established, based on performance categories. Table B shows an example of such an approach.
A typical investigation required above 100 man-hours.
The goal of this risk-based approach is to maximize value-added work, as opposed to much of the non-value-added work that would result from intensely monitoring every analytical method with a control chart. Figure 2 shows a risk-based approach for analytical monitoring.
Initially, when only small amounts of data may be available, the AMMIL should be risk-based using all available information from development and validation. Once more data becomes available, then data-driven approaches help refine the AMMIL. Monitoring should remain at level 1 when the process performance of the variable is good, such as when Ppk > 1.0.
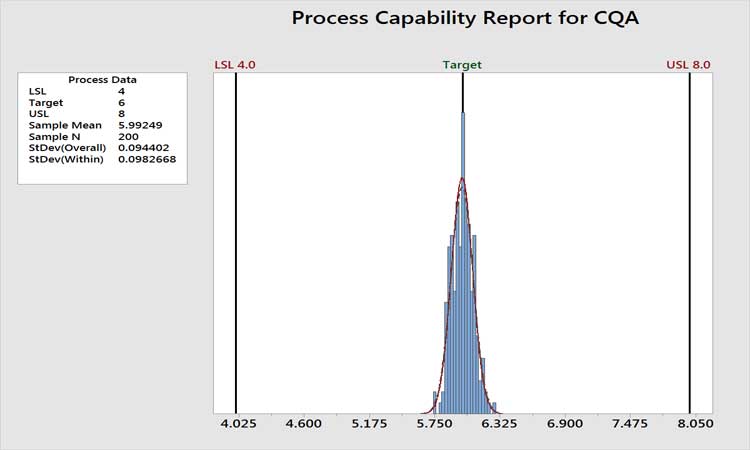
Figure 3 shows the historical performance of a variable as an example. The performance is clearly very high, with a Ppk value of around 7. The analytical method is certainly not a concern here, as variation in the measurement system cannot be greater than the total variation. In cases such as this, we do not care if the measurement system contributes 10% or 90% of the total variation. Total variation is acceptable, period. Any further work beyond level 1 AMMIL or periodic revalidation of the method would be non-value added. If the level of process performance is not acceptable, then the AMMIL should be based on a comparison of the method variability to the total variation from the process as depicted in Figure 2.
In summary, increased monitoring or improvement work for analytical methods should focus on low-performing variables where the analytical method variation is also a major contributing factor to total variability.
*Named for 19th-century Italian economist and philosopher Vilfredo Federico Damaso Pareto, the Pareto principle states that for many events, roughly 80% of the effects come from 20% of the causes.
POINT 2
The control strategy is a living business process
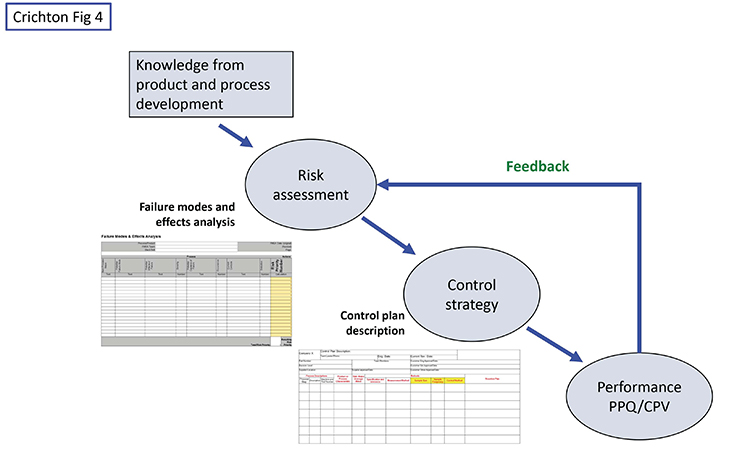
While the pharma industry is focused on risk assessments and control strategies, we believe it also has some weaknesses besides the narrow view of monitoring. One of these is too little use of formal control plans. Figure 4 shows a high-level view of the situation.
Knowledge from research and development, including subject matter expertise, experience, and design of experiments work should initially drive the risk assessment. A tool such as failure modes and effects analysis is a good device for identifying original risks before controls, and residual risks with controls in place. Starting from the original risks, the team should document how the control strategy will mitigate or control the higher risks. (Again, other industries have been doing this for decades, using the formal methodology of a control plan.) While the typical control strategy document contains most of the information, we maintain that an actual control plan document portrays the information in a clearer, more efficient, and more effective manner because it is designed to show the relationship between risks and monitoring intensity—type, location, and frequency of data gathering, with the corresponding PMIL—as well as a reaction/corrective action plan for dealing with deviations.
In our observation, the potential for false alarms (and their consequences) is generally not well understood.
POINT 3
Efficiency and effectiveness
CPV is not just a compliance program: It is a continuous improvement effort, a business process that should be evaluated as any business process would be for its efficiency and effectiveness (E&E). Companies should understand that their CPV programs add value besides compliance with FDA requirements. The agency has clearly stated that it intends for companies to learn how “[d]ata gathered during this stage might suggest ways to improve and/or optimize the process...”3 This can be accomplished by investigating signals from control charts and using historical data offline for troubleshooting and correlation studies.
Let’s take a look at the E&E of using control charts in pharma. The tendency is to want to control-chart anything and everything (just in case), highlighting signals in the statistical software and forcing many impractical investigations. So, going back to Point 1 and the word “monitor,” we need to make sure we are control-charting and reacting appropriately only where this level of intensity is required. The industry is beginning to recognize that not all signals need full investigations.4 This is a step in the right direction, but pharma needs to change risk and control plans based on performance, as discussed above.
Efficiency
Efficiency is a measure of the time and resources required to support a process and produce an output.
In the CPV context, one area of efficiency involves the amount of time and resources spent on investigations. Think about the various steps involved in a typical signal investigation:
- Record and monitor the event in a tracking system
- Investigate possible issues for this particular measurement
- Search the manufacturer’s batch records for discrepancies
- Evaluate past data
- Come up with hypothesis to test
- Declare a root cause
- Write a report that pins the root cause onto something logical
- Include any action items that result
- Send the report for review by numerous people, including quality assurance
- Potential interactions and discussions with regulatory agencies
A process control method to determine whether a variable is out of control (unpredictable versus consistent)
In thinking about efficiency, a key principle is: Every moment spent on one activity is time spent away from something potentially more important. Relating this to the Pareto Principle, it means that every moment spent on the low end of the scale is time away from the high end. Figure 5 shows the Pareto principle as applied to control chart signals. Time spent on false signals is not only costly, but takes time away from analyzing the big practical signals.
Out of curiosity, one us studied the number of false alarms that would be generated just in the tablet compression area of a site if control charts were applied rigorously to tablet weight and hardness monitoring. Given the production volumes, number of lines, etc., a 1% false alarm rate typical of the four Nelson Rules†6 was calculated to result in around 10 false alarms per day. It’s easy to see how expensive this would be.
We collected anecdotal data from knowledgeable people who perform investigations at various sites. Our findings showed that a typical investigation required above 100 man-hours on average per investigation. One site estimated the cost to be around $5,000 per investigation—a figure that we regard as extremely conservative. Now multiply this by the number of potential false alarms, and it’s easy to see why this is such an expensive proposition. It’s also why we need to focus on the business side of CPV.
In our observation, the potential for false alarms (and their consequences) is generally not well understood. Even in the simplest control-charting scenarios, applying all of the common Western Electric rules,6 as many practitioners do in statistical software, can lead to false alarm rates of 2% or more. Imagine your control chart giving a false alarm on average every 50, or even every 100, observations. Now imagine all of your control charts giving false alarms every 50–100 points. The cost implications are staggering; what’s more, consider the harmful effects of making process changes based on the spurious root causes identified for these false signals.
Since the FDA issued its 2011 guidance, the tendency in pharmaceutical manufacturing has often been to overmonitor process and product parameters
Effectiveness
Effectiveness, on the other hand, measures how well a process achieves its intended purpose.
How would effectiveness be seen in a CPV context, especially in reaction to control chart signals and the resulting investigations? Very simply, how often is a root cause actually found, a solution implemented (to either correct or prevent the cause in the future), and data gathered to demonstrate that the changes are actually working?
Every site has a favorite case study that can be pulled out of the file cabinet to demonstrate that the CPV process of investigation actually did something positive beyond compliance. But that is one out of how many investigations? Did the case study demonstrate improved performance? What effect did it have on the business?
In an informal survey of a number of sites, one of us found the effectiveness rate of investigations was estimated to be below 10%. It appears that the real motivation behind these investigational reports is to complete them on time, have a logical “excuse” for the signal, and stay out of trouble with the FDA when asked about them. After all, the cost of this inefficiency and ineffectiveness is passed on to consumers/patients/governments, so what is the incentive to change?
Part of the answer lies in the realization that the ostensible root causes found for false or minor signals are likely to be erroneous. (We can only wonder how likely is it that enacting “corrective” actions will, in fact, do harm rather than good, and inflict unacceptable societal and financial costs in the process.) Another factor that should motivate change is the increased scrutiny pharma is beginning to face from payors, regulators, and even politicians. As pricing comes under greater pressure, continuing to allow costly inefficient/ineffective practices will cease to be viable. We recommend that industry and regulators together study these issues from a more practical, realistic, and cost-conscious perspective, while, of course, maintaining focus on patient risk.
We encourage industry and regulators to adopt cpv procedures that draw on best practices from other industries.
RECOMMENDATIONS
As we mentioned earlier, it’s important to stay updated on the PMIL required of variables being monitored. One way to do this is to limit the number of control charts that require reaction plans—designate only those that are truly needed. Second, utilize the Pareto principle to focus response on practical signals, based on risk (Figure 4). Set up systems that will detect the signal as quickly and as close to the source as possible. Finding signals weeks later, when the test results become available and much more production has taken place, makes identifying true root causes extremely diffcult. It’s not unlike the criminal justice axiom that says if good evidence is not found within 48 hours, the chances of solving the crime fall dramatically. The same logic could be applied here. Other good suggestions for improving effectiveness can be found in Scherder.5
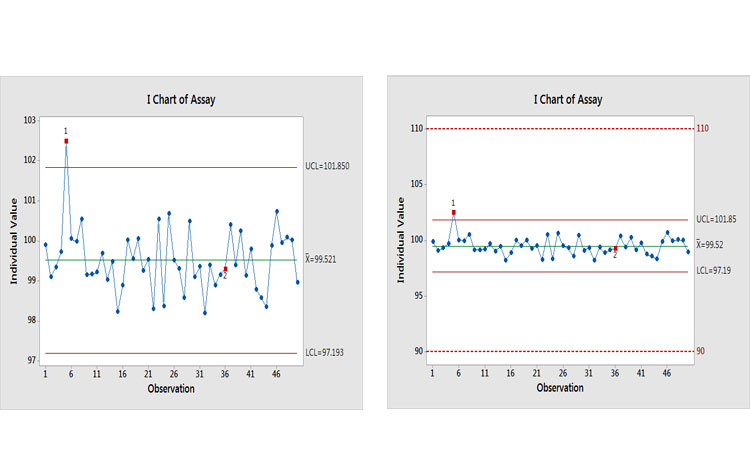
Let’s look at an example to see what the potential effects of an investigation could be. Figure 6(a) shows a control chart of an assay with two signals of potential special causes. Past thinking would suggest that investigations should be opened for each of them. This is where realism, business sense, and risk-based thinking are needed. Figure 6(b) puts this finding into context by showing it relative to specifications.
Why is this being control-charted? The performance would suggest that a lower MIL be applied. The Ppk value for this process is around 3.5, with an estimated out-of-specification (OOS) risk of <0.0000002%, which is the value when Ppk = 2.0.
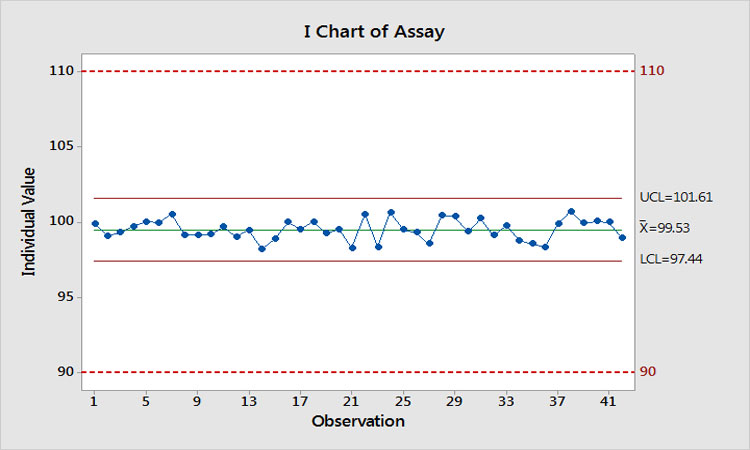
Let’s suppose we actually do find a root cause and implement a successful solution. What would be gained? Figure 7 shows what would happen. We have eliminated the special-cause variation, Ppk has increased to 3.9, and the estimated risk of OOS is now even further < 0.0000002%. Imagine when these results are presented to management to demonstrate that we just spent $10,000 on two investigations to reduce the risk of OOS from << 0.0000002% to <<< 0.0000002%!
- 5Scherder, Tara. “Embrace Special Cause Variation during CPV.” Pharmaceutical Engineering 37, no. 3 (May-June 2017).
CONCLUSION
Companies have choices in approaching CPV. In the US, the FDA has made it clear that companies can and should exercise rational discretion in adapting the degree and type of monitoring applied to a given parameter, based on the risk of deviation from required performance. MILs such as those proposed here provide a valid and practical mechanism for implementing such a program, while maintaining focus on the Pareto principle for fundamental process improvement.
Nonetheless, since the FDA issued its 2011 guidance, the tendency in pharmaceutical manufacturing has often been to over-monitor process and product parameters, even when unjustified by any realistic risk of nonconformance to specifications. In many organizations, dogma then requires investigation of any and all signals that arise. Together, these practices squander countless man-hours of effort in pursuit of process deviations that are minor or, in some cases, entirely spurious. This waste not only builds unnecessary cost into all pharma products thus affected, but actually incurs risk by diverting resources from more to less critical opportunities for improvement and by raising the possibility that some of the “fixes” applied may actually result in harm rather than good.
The authors wholeheartedly endorse a risk-based approach to process monitoring that embraces the continuum paradigm the FDA has articulated, and which employs the MIL concept for both processes and analytical methods to implement a “statistically appropriate and representative level”3 of oversight for each key parameter. Such methods are truly customer-focused and provide the means to maximize efficiency and effectiveness of the business process, while maintaining and improving quality.
We encourage industry and regulators to work more closely to adopt CPV procedures that draw on established research and known best practices from other industries, focusing on substance rather than form, to improve quality, reduce costs, and promote the public good.


