Repurposing a US Cell Therapy Facility for Flexibility and EU Compliance

The explosive growth of advanced therapy medicinal products (ATMPs), particularly cellular therapeutics, has driven steady investment in facilities capable of manufacturing these therapeutics at scale . Meanwhile, the industry is collectively moving to adapt to European Union Annex 1 standards, which places a more stringent emphasis on contamination control.
In this conceptual design case study, we discuss plans to convert an existing single-product cell therapy facility into a contract development and manufacturing organization (CDMO) facility. This facility would be capable of running multiple lines of batchedbased therapies while maintaining GMP compliance with EudraLex Volume 4, Annex 1 and the guidelines on GMP for ATMPs.3,4 The new CDMO-focused portion of the facility will encompass 25,000 square feet of an existing 42,000-square-foot manufacturing area. It could be converted while the remaining space continues existing operations.
| Potential Risk | Engineering Considerations |
| Handling of viral vectors in a multiproduct/multiclient facility could lead to cross contamination. | Design for dedicated, single-pass heating, ventilation, and air conditioning (HVAC) systems to ensure that the air in any given production suite is not recirculated back to other areas. |
| Use of pressure cascades in the production suites could lead to cross contamination in a multiproduct/multiclient facility. | Design airlocks with a bubble/sink configuration final to protect the manufacturing environment and outside areas. |
| In a multiproduct/multiclient facility, final product, apheresis, equipment, etc. may be taken through a given area at the same time with the potential for contamination. | A strictly engineering solution cannot be implemented. This will require adequate procedures to ensure segregation of materials and personnel as needed. |
| The multiproduct/multiclient facility will have gases and water supplied to the suite with piping. Cleaning piping can be a challenge and could lead to contamination. | Design the piping to include easily cleanable covers and, when possible, recess the piping into the ceiling so that removable flex classified connections can be made. |
| Currently, the area transitions from controlled not classified (CNC) to Grade C. This increases the risk of regulatory observation and the potential for contamination. | Design the airlocks so that there is progression from CNC to Grade D, to Grade C, to Grade B. |
| Multiple batches may be stored in an incubator at the same time, increasing the risk of cross contamination. | • Engineering: Fully exhaust air from the incubators where this will happen. • Procedural alternative: Ensure segregation of batches within the incubator. |
| Manufacturing multiple batches in separate biosafety cabinets (BSCs) or isolators in the same room can lead to cross contamination. | • BSCs engineering: Fully exhaust air from any BSC in the room where multiple batches are being processed. • Procedural alternative: Ensure physical segregation of batches and dedicated operators within the room. • A BSC risk assessment must be conducted by the company to show why using multiple BSCs at the same time is acceptable, with or without BSC exhaust. • Isolators are the preferred engineering solution because they provide a high degree of assurance that the risk of cross contamination is reduced. |
The transition of cell and gene therapies (C>s) from laboratory to clinical use has been a revolution, decades in the making5,6. ATMPs are poised to capture a significant portion of the biopharmaceutical industry, which motivated the subject company of this case study to expand its current business model to incorporate CDMO practices 1.
FACILITY CONVERSION DESIGN
The approaches to facility conversion design presented in this case study can serve as a model for repurposing existing manufacturing space for ATMP processes while adhering to the revised Annex 1 standards implemented as of 2023. Here, we provide an overview of the airflow and room classification modifications necessary to make this facility compliant with EU regulations for concurrently manufacturing multiple products. As part of the initial concept effort, risks were reviewed and addressed or identified and documented so that they could be further analyzed and addressed in later stages of the design (see Table 1).
ATMP OPPORTUNITIES AND CONSIDERATIONS
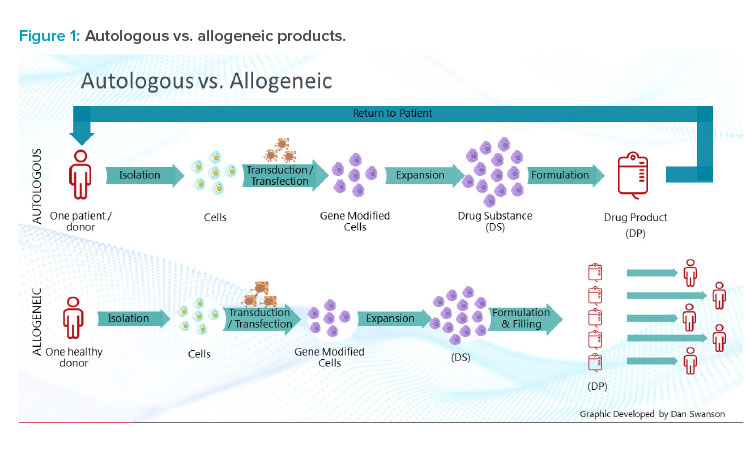
ATMPs encompass several cell- and tissue-based techniques, including in vivo and ex vivo gene and somatic cell therapies 6. Cell therapies can be either autologous, which involves harvesting, manipulating, and administering modified cells back to the original donor patient, or allogeneic, which involves cell-based therapeutics derived from donated blood or tissues that are expanded at a much larger scale to enable treating multiple patients (see Figure 1) 7.
The transformative chimeric antigen receptor T cell (CAR T) therapy, for example, starts with a patient’s own T cells, which are isolated from an autologous donation of blood or leukapheresis product (leukocytes or white blood cells isolated from blood). The donor blood or leukapheresis product is collected in a manufacturing facility in a closed IV bag known as a leukopak. Then, it is shipped to a manufacturing facility where the patient’s T cells are modified to produce CARs. The CAR T therapeutics specifically target and destroy cancer cells, rendering tumors vulnerable to the patient’s immune system 8,9.
CAR T products fall into two different types: autologous products and allogeneic products. Understanding which products will be processed will heavily inform facility design. Autologous products focus on a single patient and/or donor, with all products being produced specifically for that patient. Allogeneic facilities have the potential to be much more efficient by generating larger batches for administration to a wider patient population.
In this facility, the company plans to focus on contract autologous CAR T processes, with the flexibility and capacity needed to scale-up CAR T and natural killer allogeneic processes.
Autologous therapeutic manufacturing presents a sizable challenge for GMP production at scale, which involves scaling out additional copies of the process, regardless of open or closed format7. In this mode of operation, increased batch turnover presents more opportunities for batch mix-up and cross contamination to occur. In addition, complex personnel flows, material flows, and higher batch throughput required to meet the demands of growing clinical and commercial programs increase the opportunity for microbiological contamination 10.
THE CHALLENGES OF COMPLIANCE WITH EUROPEAN REGULATIONS
Contamination control is a fundamental focus of the revised Annex 1 regulations. Although Annex 1 and the guidelines on GMP for ATMPs are specifically meant for therapeutics developed for European markets, they also represent a new gold standard for modern GMP across the industry. The US Food and Drug Administration (FDA) has not only taken notice, but has also helped contribute to defining Annex 1 standards. Annex 1 impacts a facility’s design if even one product made on-site is intended for European markets.
Annex 1 requires manufacturers to develop a contamination control strategy to govern their manufacturing process, which may require a comprehensive reexamination of processes at a given facility3.
One of the driving factors in the new Annex 1 regulations is the contamination control strategy, including the prevention of cross contamination.
The simple solution is often new construction, which allows a fresh start with each new product line. However, the advent of ATMPs, benchtop processing equipment, and bioreactor-based manufacturing using single-use components has brought about new, beneficial economies to batch-based manufacturing 8. These processes allow a single facility to produce a continuously changing array of new products without dramatically altering the manufacturing space. If the correct array of process utilities and adequate capacity are in place, manufacturers can leverage the same space and the same or similar benchtop bioreactors and equipment for different processes.
Mobile lab bench configurations and modular equipment enable equipment changes to support a range of client processes using different brands of equipment platforms. The question then becomes whether the facility, flows, engineering controls, and associated procedures can become robust enough to continuously change production while adhering to the current GMP.
In this case study, an existing cell therapy manufacturer leverages extra capacity, physical space, and in-house expertise in ATMP manufacturing for contract production as a CDMO. The company’s intent was to take a facility nearing the end of its life cycle and modify it to provide new opportunities for growth and revenue streams.
OVERVIEW OF PROPOSED MODIFICATIONS
Built over a decade ago, the 180,000-square-foot facility remains a relatively robust, state-of-the-art ATMP manufacturing facility. The site was strategically built near a major transportation hub to expedite shipments of autologous cell products quickly and efficiently. The manufacturing floor consists of six Grade B production modules. Each currently houses multiple cell therapy processing workstations, including BSCs, incubators, centrifuges, and other technical equipment.
Upon completion of the project, most of the process (from donor material thaw to formulation and fill) will be contained within the individual production modules. The warehouse and product freezing/storage room will house controlled-rate freezers to cryopreserve intermediates and final products, as well as liquid nitrogen freezers to store incoming and outgoing materials. Liquid nitrogen freezers in the warehouse may also be used for long-term storage of master cell banks or working cell banks for allogeneic processes.
In the conversion, the owner intends to use four of the modules as a GMP manufacturing area that will initially produce cell therapy products for Phase I/II/III clinical trials. This will include the ability to support commercial manufacturing for select clients. The facility is currently designed primarily for autologous CAR T processes with built-in flexibility and capacity to accommodate allogeneic therapy manufacturing. The typical production processes within the Grade B modules are expected to take 7–14 days, depending on the nature and scale of the client’s process. The remaining two modules will continue to manufacture the company’s flagship ATMP.
One of the driving factors in the new Annex 1 regulations is the contamination control strategy, including the prevention of cross contamination. The new CDMO space will be a multiclient and multiproduct facility, which will require changes to how people and materials will move throughout the facility. Changes to room classifications and air pressure cascades will be implemented to comply with Annex 1 guidelines on containing potential contaminants.
The designed CDMO area of the facility contains the four identical Grade B production modules in addition to support spaces. These include areas for parts prep, product freezing and storage, labeling and inspection, and waste-out staging; a dispensary; chemistry, manufacturing, and control (CMC) stability rooms; quality control labs; a warehouse; and utility rooms.
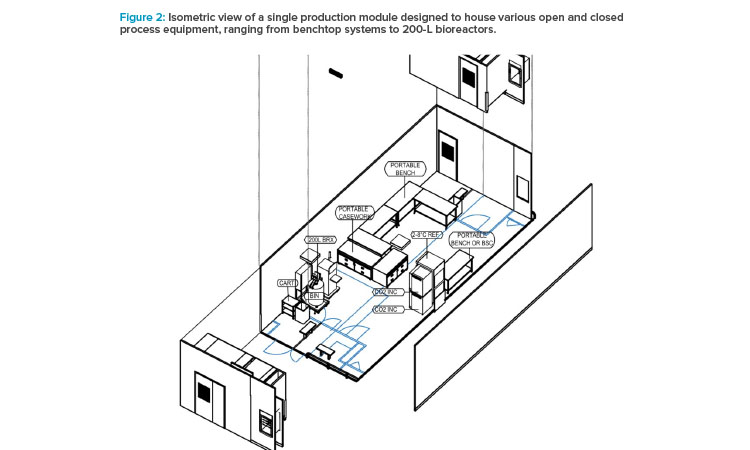
The concept design for the production modules includes flexibility to meet future client processing needs with the capability to house equipment for open or closed processing. Meanwhile, the rooms are operated under Grade B or Grade C backgrounds, respectively (see Figure 2). Design features include portable benches and process gas utility panels placed in flexible locations to accommodate equipment changes. Process utilities are sized accordingly to support a range of cell culture vessels up to 200 liters (L).
To evaluate and determine the appropriate layout of the updated production modules, we performed a capacity analysis of various production module configurations. These included autologous open processing, autologous closed processing, and allogeneic closed processing at a concept design level. For both autologous and allogeneic closed process configurations, two equipment arrangement scenarios were considered. The tradeoffs of varying the number of closed process systems (such as Miltenyi Biotec CliniMACS Prodigy, benchtop bioreactor, and 200-L bioreactor systems) were weighed (see Table 2).
Three facility operational scenarios were also evaluated to study varying the number of autologous vs. allogeneic process modules and the impact on the facility production capacity. At a concept design level, we produced layouts of all configurations described in Table 1 and sized process utilities to support two modules with bench-scale reactor processes and two modules with 200-L bioreactors. A GMP equipment storage room has also been designed to accommodate turnover of the different module equipment configurations based on CDMO client need.
| Configuration Description | BSC Quantity | Closed System Quantity | Bioreactor Quantity (Benchtop) | Bioreactor Quantity (200 L) | Module Quantity | Total Capability (Number of Concurrent Lots |
|---|---|---|---|---|---|---|
| Operational Scenario 1 | ||||||
| Autologous - Open Process | 6 | - | - | - | 1 | 6 |
| Autologous - Closed Process Equipment Scenario 1 | 2 (inoculation/fi ll) | 6 | - | - | 1 | 6 |
| Autologous – Closed Process Equipment Scenario 2 | 2 (inoculation/fi ll) | - | 6 | - | 2 | 12 |
| Total CDMO capability (number of concurrent lots | 24 | |||||
| Operational Scenario 2 | ||||||
| Autologous - Closed Process Equipment Scenario | 2 (inoculation/fi ll) | 6 | - | - | 2 | 12 |
| Autologous - Closed Process Equipment Scenario 2 | 2 (inoculation/fi ll) | - | 6 | - | 1 | 6 |
| Allogeneic - Closed Process Equipment Scenario 1 | 3 (inoculation/fi ll) | - | 6 | - | 1 | 6 |
| Total CDMO capability (number of concurrent lots | 24 | |||||
| Operational Scenario 3 | ||||||
| Autologous - Closed Process Equipment Scenario 1 | 2 (inoculation/fi ll) | 6 | - | - | 2 | 12 |
| Allogeneic - Closed Process Equipment Scenario 2 | 3 (inoculation/fi ll) | - | 2 | 2 | 2 | 8 |
| Total CDMO capability (number of concurrent lots | 20 | |||||
Considering that the four modules being repurposed for the CDMO function were fully staffed to produce the company’s ATMP product, this conceptual study assumes a similar level of staffing and material storage requirements to the original operation.
Future design phases will leverage industrial modeling expertise to build detailed simulation models that will verify the following based on the final suite configuration and output required by the company: equipment quantities, staffing levels, scheduling, material movements, storage requirements, and total facility output.
The guidelines require technical and organizational measures to separate the activity, which concerns the fl ow of people and material through the processing suite.
Additional operational considerations such as multiple shifts, media prep, and cold room storage spaces shall also be considered and modeled during future design phases. Based on the number of 200-L processes chosen for the final facility layout, automated filling and inspection equipment to support large-scale allogeneic processes must be carefully considered. Isolated filling lines can be operated in a Grade C or Grade D background, reducing the required Grade B footprint 3. Semi- or fully automated inspection machines can be operated in CNC areas and offer the benefits of a lower inspection footprint and lower staffing levels.
The construction phase will update supporting spaces for Annex 1 compliance. This includes directional airflows, ensuring airlock doors are interlocked, implementing active pass-throughs, and installing windows or cameras to allow visibility into production suites. The individual production modules will also be converted to comply with EU regulations for multiclient and multiproduct use as outlined in the guidelines on GMP for ATMPs. This includes the design of segregated areas for specific process steps, use of airlocks with pressure sinks and bubbles to confine potential airborne contaminants within a specified area, use of closed systems, and the use of single-use technology.
FACTORS FOR COMPLIANCE
On paper, the Annex 1 revisions and existing guidelines on GMP for ATMPs seem vague on requirements for a multiproduct ATMP facility. For that reason, it helps to keep the overall intent of the regulations in mind while interpreting them for specific circumstances—the prevention of mix-ups and cross contamination for the safety of the patients who will ultimately receive the therapeutic.
The guidelines require technical and organizational measures to separate the activity, which concerns the flow of people and material through the processing suite. One dramatic shift in the latest Annex 1 revisions clarified requirements for the transition between areas of different classifications. This allows only a single step up between classified spaces. For example, you could pass through an airlock between a Grade C and Grade B space, but not from Grade D to Grade B 3.
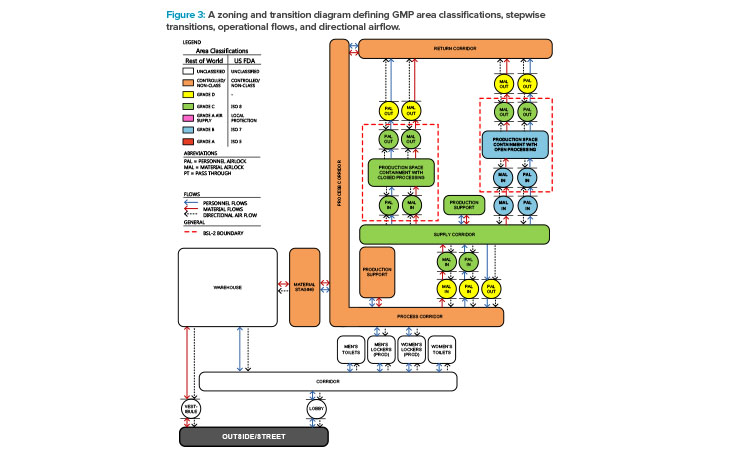
This requires unidirectional people and material flows. This begins with a CNC space, an area that meets a company-defined criteria for entry into classified areas or where materials and personnel may traverse under controlled conditions outside of the classified environments. It then transitions through Grade D and Grade C spaces to enter the Grade B manufacturing modules (see Figure 3). People and materials move through a series of sink (negative air pressure) and/or bubble (positive air pressure) airlock transition spaces, where adjacent rooms of different grades have a pressure differential designed to better contain contaminants and viral vectors.
The Grade B modules will be used to manufacture biosafety level 2 (BSL-2) products, as the manufacturing processes use human cells and lentiviral vectors. Although there are no prescriptive regulations on directional airflow for BSL-2 processes, the Centers for Disease Control and Prevention (CDC) and National Institutes of Health (NIH) publication on biosafety in microbiological and biomedical laboratories recommends inward airflow and no air recirculation to spaces outside of the BSL-2 boundary when considering BSL-2 containment11.
We considered this recommendation in the context of the updated multiproduct and multiclient facility and implemented a containment design featuring a bubble entry airlock and a sink exit airlock to prevent contaminants from entering or escaping, respectively.
The design establishes the rules that the layout should follow. To meet Annex 1 requirements for pass-throughs and stepwise transitions, existing material pass-through hatches between CNC/Grade C and Grade C/Grade B spaces must be converted to dynamic pass-throughs. This means the flow of objects through pass-throughs requires HEPA air filtration to allow the passage of material but not airborne contaminants. In addition, material airlocks from CNC to Grade C should be classified as Grade D, and pass-through hatches for materials entering Grade B production modules should be classified as Grade B to comply with Annex 1.
The guidelines on GMP for ATMPs also call for single-pass air for areas handling multiple viral vectors or for multiproduct suites. Conceivably, it is possible to recirculate HEPA-filtered air in suites devoted to a single product. Still, a contamination strategy would need to prove, with evidence, how the entire HVAC system would be decontaminated4.
The design basis is to allow each module to continue operating independently from other air handling unit (AHU) systems, enabling the modules to be upgraded at different times. This would require separate AHUs for each module. To meet multiproduct facility requirements for single-pass air listed in the guidelines on GMP for ATMPs, the current AHUs must be replaced with higher-capacity systems, which will also require additional modifications to utility supply lines and additional capacity to the current facility’s chiller plant.
CONCLUSION
Upgrading an existing facility to meet the regulations outlined in Annex 1 and the guidelines on GMP for ATMPs involves complex decisions regarding HVAC and the flow of people and material. The facility’s original layout allowed for cordoning off CNC spaces and room for the stepwise transition between classified spaces. Air handling and filtration will require significant capital costs to reconfigure the manufacturing space to accommodate the needs of a multiproduct CDMO.
Those costs, however, need to be weighed against the expense of developing new facilities from scratch. As ATMP technology matures, the demand for C> product manufacturing space will only increase. This brief example shows the significant considerations for converting a facility from a single ATMP product manufacturing to multiproduct manufacturing following a more stringent regulatory framework. It also demonstrates that the conversion is possible and that it may help bring these important products to the markets faster than a new greenfield facility.


