CMC Requirements for New Drug Registration in Latin America

The global pandemic has demonstrated that now, more than ever, we need to work toward a global solution and prioritize the harmonization of technical requirements. Positive improvements have been observed in the acceptance and implementation of international standards by various regulatory agencies in Latin America. This article offers an overview of the chemistry, manufacturing, and controls (CMC) requirements for the small molecules product registration process in Latin America and highlights the divergence of some requirements from harmonized standards like the International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH).
In an era when the world is accelerating the development of drugs and targeted medicines using innovative technologies, pharmaceutical companies still face registration hurdles for well-characterized molecules because of redundant or additional local regulatory requirements. There is a concerted effort from multinational pharmaceutical companies in developing countries to embrace regulatory convergence with international standards, primarily ICH, and to implement these standards to meet the increasing needs for and to expedite patient access to specialized and targeted medicines.
Improvements have been observed by various regulatory agencies in Latin America for the acceptance and implementation of international standards—for instance, the ICH Common Technical Document (CTD) format. However, existing, or new local regulatory requirements still present hurdles and the regulators’ intention to embrace ICH guidelines or to accept alternate risk-based, scientifically supported approaches may be interpreted negatively by global pharmaceutical companies.
When companies pursue drug registration in these markets, the need for additional country-specific documents—such as certificates of pharmaceutical products (CPPs), Good Manufacturing Practice (GMP) certificates, site master files, and various types of declarations—may deter or delay progress and prevent timely patient access to drugs. These various local regulations inhibit the pharmaceutical industry from achieving its goal of harmonized international guidance documents and a single global dossier with harmonized terminology, regulatory standards, and evidence requirements.
Alternate approaches to meeting local requirements emerged during the COVID-19 pandemic: one example is the acceptance of electronically generated regulatory documents (e.g., CPPs). Regulatory agencies can learn from the transformative experience required by COVID-19 pandemic restrictions. And beyond that, they should willingly improve regulatory frameworks to streamline processes and global harmonized requirements (e.g., ICH) and to consider the adoption of reliance procedures that benefit patients via faster access to quality medicine.
This article offers an overview of the CMC requirements for the small molecules product registration process in Latin America. The information results from the authors’ experience working in a global company and managing successful drug registrations in the Latin American region. The Latin American region defined here includes the following Central and South America countries: Argentina, Bolivia, Brazil, Chile, Colombia, Costa Rica, the Domini-can Republic, Ecuador, El Salvador, Guatemala, Honduras, Mexico, Nicaragua, Panama, Paraguay, Peru, Uruguay, and Venezuela. Caribbean markets are excluded from this analysis.
Regional Registration Processes
In the absence of a harmonized regional process for drug registration in Latin America, local regulations and processes set by each individual country are established. Some of these are more stringent than ICH requirements (e.g., Brazil requires specific forced degradation studies and analytical validation requirements)1 and require more detail, which may ultimately restrict flexibility for drug supply, at initial registration, and during product life cycle management related to postapproval changes.
In the past few years, improvements in regulatory systems have been observed in Latin America, showing a stepwise harmonization of local regulations with international standards such as ICH. Despite the complex and challenging process of creating or reviewing a regulation in the region, many draft and new regulations have been published, illustrating regulatory agencies’ willingness to improve their regulatory frameworks. A few examples of new or revised regulations include the Chile Institute of Public Health (ISP) adopting the CTD format for Module 3 for new drug registration,2 creating new timelines for agency review and approval of regulatory submissions, and the Colombian Ministry of Health creating new timelines for agency review and approval of regulatory submission and publishing newly established reporting categories for postapproval changes.3
Food and Medical Technology/Administración Nacional de Medica-mentos, Alimentos y Tecnología Médica) in Argentina, CECMED (Regulatory Authority for Medicines, Equipment and Medical Devices of the Republic of Cuba/Autoridad Reguladora de Medicamentos, Equipos y Dispositivos Médicos de la República de Cuba) in Cuba, and INVIMA (National Institute for Food and Drug Surveillance/Instituto Nacional De Vigilancia de Medicamentos y Alimentos) in Colombia. This means that the registration process in these markets depends heavily on local regulations.
In the absence of a harmonized regional process for drug registration in Latin America, local regulations and processes set by each individual country are established.
Note that a country’s membership in ICH does not translate into immediate adherence to the guidelines: ICH implementation plans are used generally to determine the transition. However, implementing ICH guidelines in these markets requires additional time and resources to both properly interpret the guidelines and to work them into local rules by revising, creating, or eliminating regulations.
Until full transition is achieved, even the two ICH members (Brazil and Mexico) must still use local redundant documents for initial drug registration. Latin American countries widely recognize the US and EU as reference markets—or country of reference (COR)—as part of the drug registration process. However, drug product approval in a COR doesn’t necessarily mean local registration requirements are harmonized with dossiers submitted and approved in the COR, or that the local drug review process is accelerated as a result of the COR. The request for far more data than originally required or approved in the COR should be anticipated, including where the data are generated (e.g., the site where development occurs versus where commercialization is intended). Additional required information beyond ICH guidelines, or non-value-added documents (e.g., declarations), require companies to prepare and maintain multiple versions of the registration dossier across the region. These situations can lead to filing and approval delays for new medicines and can create additional burden on product life cycle management.
Companies often must manage divergent and complex regulatory requirements due to differences in local postapproval regulations and requirements since additional registration submissions are required for low-risk changes. This, in turn, can lead to drug stock outs and shortages. One example of such complexity is that Latin American health authorities cannot accommodate minor changes that require a simple notification in US4 and/or EU5 (the “do and tell” procedure) but are considered instead a major change (e.g., Colombia)6 that requires a minimum of 6- to 12-month review and approval timeline.
Country-Specific Registration Requirements
Despite the evolving growth of local drug regulations and increased global harmonization efforts in Latin America, it is reasonable to state that the registration process is still highly country-specific. Although Latin American regulators seek global harmonization, they struggle with the right balance of local versus global requirements, which leads to an increased demand for country-specific documentation.
Table 1: Appendices and drug substance Module 3 required sections
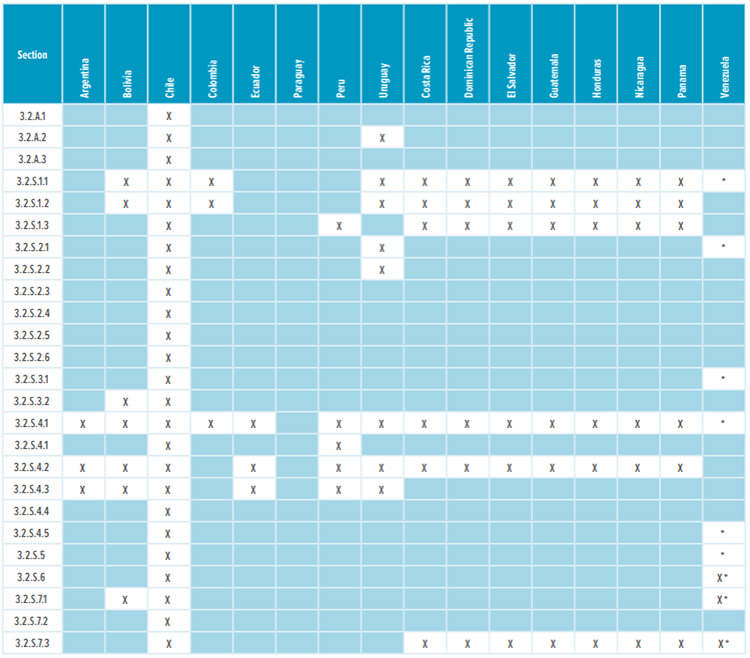
* Corresponding Quality Overall Summaries (QOS) sections 2.3.S.1, 2.3.S.2, 2.3.S.3, 2.3.S.4, 2.3.S.5, 2.3.S.6, and 2.3.S.7 are required.
Most countries in Latin America, except Brazil and Mexico, do not require a full Module 3 CTD dossier to register a drug. Table 1 and Table 2 list the CTD sections that are needed in the registration dossiers where a full Module 3 is not required. The content of regional sections is not listed because this section differs significantly from country to country. Depending on the country, dossiers require CMC/Quality documentation that isn’t usually included in the global CTD dossier. Examples of such documentation include:
- Certificates of analysis (COAs) of all formulation components from both the drug product manufacturer and supplier
- Executed batch records, including packaging records
- Chromatograms from analytical testing for batches on stability
- Analytical validation protocols and reports
- Specific stability information provided in a specific format such as statements, declarations, and memos
- CTD sections signed by the manufacturer, even when the manufacturer is not necessarily the marketing authorization holder
- Local testing/release
This article discusses the most common requirements that may impact dossier preparation and registration: approval in COR/country of origin (COO), CPP, stability studies, ancillary documents, and other requirements.
Table 2: Drug product Module 3 required sections
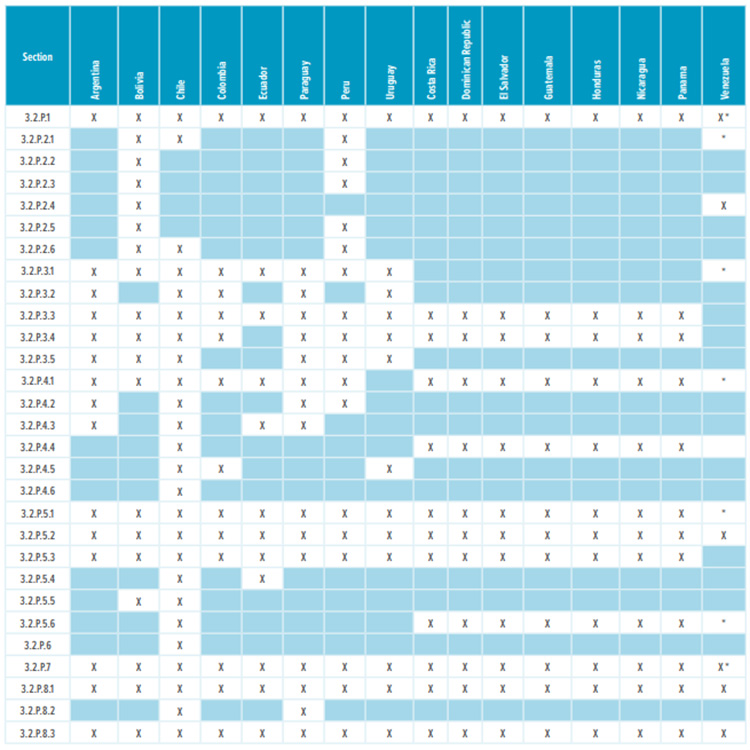
* Corresponding Quality Overall (QOS) sections 2.3.P.1, 2.3.P.2, 2.3.P.3, 2.3.P.4, 2.3.P.5, 2.3.P.6, and 2.3.P.7 are required.
Approval in Country of Reference (COR)/Country of Origin (COO)
The drug registration process in Latin American countries depends heavily on first approval in a well-recognized country with competent regulatory agency, known as the COR. Therefore, the US and EU are widely recognized as COR markets. The fact that the US and EU follow ICH guidelines does not really dictate the registration process, nor does it drive the requirements for a harmonized registration process in Latin America.
In addition to the COR, some Latin American markets require approval in the COO, which is defined as the country where the drug is manufactured, packaged, or exported from. Therefore, when planning submissions in a specific market in Latin America, it is important to take this requirement into account.7 A review of COR/COO requirements in Latin America shows that most markets mandate that the product is first approved in the COR—and it is tied to the availability of a CPP to be able to submit a new marketing application and to the product being approved in the COO before submission or approval.7
| Manufacturing License | GMP Certificate | CPP | |
|---|---|---|---|
| Argentina | √ | √ | √ |
| Bolivia | √ | √ | √ |
| Brazil | √ | √ | √* |
| Chile | Not required | √ | √ |
| Colombia | √ | √ | √ |
| Costa Rica | Not required | √ | √ |
| Guatemala | Not required | √ | √ |
| Honduras | Not required | √ | √ |
| Mexico | √ | √ | √ |
| Panama | Not required | √ | √ |
| Peru | Not required | √ | √ |
| Venezuela | Not required | √ | √ |
* Approval letter accepted in lieu of a CPP; not required at the initial submission but before ANVISA approval.
| Countries | Climatic Zone* | API Stability Required? (Y/N) | In-Use Stability Required? (Y/N) | Photo-Stability Required? (Y/N) | Minimum Required Data at Submission |
|---|---|---|---|---|---|
| Argentina | II | N | N | N | 6 months accelerated / 12 months long term |
| Bolivia | IVA or IVB | Y | Y | N | 6 months accelerated / 6 months long term |
| Brazil | IVB | Y | Y | Y | 6 months accelerated / 12 months long term |
| CAC** | IVB | N | Y | N | 6 months accelerated / 12 months long term |
| Chile | II or IVA | Y | Y | N | 6 months accelerated / 6 months long term |
| Colombia | IVB | N | Y | N | 6 months accelerated / 12 months long term |
| Ecuador | IVA or IVB | N | Y | Y | 6 months accelerated / 12 months long term |
| Mexico | II | Y | Y | Y | 6 months accelerated / 12 months long term |
| Paraguay | IVA or IVB | N | Y | N | 6 months accelerated / 12 months long term |
| Peru | IVA or IVB | N | Y | N | 6 months accelerated / 6 months long term |
| Uruguay | II | N | N | Y | 6 months accelerated / 6 months long term |
| Venezuela | IVB | Y | Y | N | 6 months accelerated / 6 months long term |
* II: 25°C/60% RH; IVA: 30°C/65% RH (hot and humid); IVB: 30°C/75% RH (hot and very humid)
** Central American countries (Costa Rica, Dominican Republic, El Salvador, Guatemala, Honduras, Nicaragua, and Panama).
Certificate of Pharmaceutical Product (CPP)
The CPP is intended to facilitate regulatory review and to replace a full dossier evaluation of the quality, safety, and efficacy of the requesting country. When effectively used, this accelerates approval and early patient access to innovative medicine. The CPP is required in Latin American regions to support a regulatory submission at the beginning of or during the health authority review. According to the World Health Organization (WHO) Scheme, CPPs should not be required in countries that require full ICH CTD dossiers and that have the capability to conduct full quality, safety, and efficacy reviews.8
When developing a regional submission strategy for Latin American countries, CPP requirements are considered early in the planning phase. If required, national regulatory authorities should be open to discussion in advance of the regulatory submission to give advice and agree on the content of the submission, including the CPP, to move forward as quickly as possible.9, 10
In practice, all Latin American countries conduct detailed evaluations of drug registration applications, even if they require and receive a CPP. For markets with fast-track, simplified, and/or reliance procedures (e.g., Argentina,11 the decision is based on the recognition of certain regulatory authorities and not on the CPP. In these markets, the registration procedure for small molecules medicines is quite simple and requires minimum CMC information. For example: the approval process in Argentina is short (4 to 6 months).
Most Latin American countries do not require full CTD dossier evaluation (see Tables 1 and 2) but do require a CPP. These markets will perform a thorough dossier assessment of the submitted information with follow-up questions and information requests to applicants. Moreover, because the CPP confirms GMP status, additional GMP certificates should not be necessary. Table 3 describes the markets that require both documents (CPP and GMP certificates). Given that the CPP is a legal document, additional certification and/or legalization should not be requested. This is not what is currently observed as the CPP document is either certified or legalized.
Stability Studies
Latin America is a diverse region with different climatic zones. For instance, Chile, Argentina, and Mexico are Zone II markets (25°C/60% RH), whereas Brazil and Paraguay are Zone IVB markets (30°C/75% RH). Despite that, the most restricted climatic zone (i.e., Zone IVB) when available, is usually provided to all Latin American market registration submissions, considering the stability of the product and the proposed shelf life. Table 4 describes a high-level overview of the stability requirements in Latin America.
Three drug product stability batches are required for initial registration and ICH Q1A (R2) Stability Testing of New Drug Substances and Products12 is largely accepted in the region. Brazil, Costa Rica, Dominican Republic, El Salvador, Guatemala, Honduras, Nicaragua, Panama, Mexico, and Peru accept stability data from pilot/development batches manufactured at development sites, whereas the other Latin American countries, except Argentina,11 require stability data from the proposed commercial sites. Additionally, Mexico requires real-time commercial stability data to grant the drug product shelf life if the development site is not being registered as part of the drug product commercial supply chain. Also, COFEPRIS in Mexico doesn’t allow extrapolation of shelf life considering the long-term,real-time commercial stability data that are amenable to statistical analysis, which is allowed by ICH Q1E, Evaluation of Stability Data.13
Although the principles of ICH Q1A (R2) are widely accepted and recognized by most Latin American health authorities, some markets still require a significant customization of the stability data by means of stability declarations and other ancillary documents: these additional requirements are discussed in the next paragraphs.
Ancillary Documents and Other Requirements
Although some ancillary documents are needed to support drug registration even in a well-established market, some ancillary documents in Latin American markets go beyond what would be expected for inclusion in the dossier and sometimes these documents contain redundant information already covered in the CTD sections. Ancillary documents required to support registration in these markets include COAs, GMP certificates, test chromatograms, a batch numbering system, stability declarations, and signed declarations.
Certificates of Analysis (COAs)
Drug product COAs are required in most Latin American markets. Sometimes, COAs for the drug substance, the excipients used in the formulation, and the primary and nonfunctional secondary packaging material are requested. Most markets accept COAs from the registration batches (pilot or commercial scale) or commercial batches, if available. In Mexico, COAs from development batches can only be accepted if the site where the product was manufactured is also registered for commercial supply. In Central American markets, COAs of the samples submitted for testing are needed.
Sample Requirements
Samples of the finished product and/or standards are required in some markets in Latin America at or during the marketing application review. The challenges with providing samples are around their availability, arranging for their shipment, and preparing the appropriate regulatory documentation for custom clearance. Finished product samples are required in the following countries: Uruguay, Panama, Bolivia, the Dominican Republic (only product photos required for fast-track pathway), Guatemala (not required for fast-track pathway), and Nicaragua (for presentation only).
Compendial Monographs
Most formulations use common ingredients (excipients) and most of these excipients are of compendial grades. Additionally, quality control testing of these compounds is conducted using standard testing following applicable pharmacopeia—such as United States Pharmacopeia (USP), European Pharmacopeia (EP), and Japanese Pharmacopeia (JP)—that was current at the time of testing.
Registration dossiers in most Latin American markets require copies of the pharmacopeial monographs. Although copies are included in the dossier, they may no longer be valid when reviewed by the regulatory authorities due to ongoing updates to the compendia. Countries that require copies of excipient monographs and/or general testing monographs include Bolivia, Brazil, Costa Rica, Ecuador, El Salvador, Guatemala, Mexico, Nicaragua, Panama, Paraguay, and Venezuela.
Miscellaneous Declarations
Some of the documents that are required in the initial registration dossiers in Latin America include various declarations that serve different purposes.
Batch numbering system declaration or memo: The batch numbering system declaration or memo is a document that describes how the drug product and sometimes the drug substance batches are numbered. This information should be produced by the manufacturing site and should describe how the lot numbers are issued and assigned. The batch numbering system declaration is required in Bolivia, Chile, Ecuador, Panama, Paraguay, and Peru.
Signature declarations or memos: Although the principles of ICH Q1A (R2) are widely accepted and recognized by most of Latin American health authorities, some markets still require a significant customization of the stability data, by means of stability declarations and other ancillary documents. The declarations/memos are required individually for the drug product and reconstituted drug on stability as applicable. The information required in these declarations include product information (name, strength, dosage form), manufacturer, packager and license holder, and drug product manufacturing site name and country. Most important, this documentation also includes stability batch information such as batch size, type, manufacturing and expiry dates, packaging configurations, start and end date of the stability studies, analysis dates, storage conditions, quantities of samples on stability, and conclusions with specific wording. Sometimes the stability chamber type and the material and the color of the container closure system are required. Signature memos are required in Peru, Ecuador, and Central American markets. Additional declarations to describe how the drug is described/referred to in the CTD components (generic molecule name/code) versus the proposed trade name or how a test result is reported on the COA versus how it is described in the specification (e.g., how the attribute “appearance” is described in the specification document specifically for pass/fail or complies type of report-ing) all need to be explained. These declarations are needed in Central American markets.
The key message spread during the pandemic is that the industries and regulators should apply the learnings from this unprecedented global experience to streamline their current processes and seek opportunities for greater international collaboration.
Chromatograms: The chromatograms for testing methods performed by high performance liquid chromatography, gas chromatography, or infrared spectroscopy are required for drug substance (Chile, Mexico) and/or drug product (Ecuador, Mexico, and Uruguay). The chromatograms for the drug product stability testing from the initial analysis (time zero) and all subsequent time points—at least for the identification, assay, and dissolution tests— are required for all registration batches on stability tested under long-term (Ecuador, Mexico) and accelerated conditions (Mexico). The chromatograms must include the blanks and standards (Mexico) and must be clearly identified with the batch number, the time point, and storage conditions. The chromatograms must show date, time, volume of injection (Ecuador, Mexico), and the area under curve (Mexico). The same is required for the drug substance in Mexico. In Uruguay, the certificate of analysis of a recent manufactured lot of a drug product with its chromatograms or spectrums (sample, reference standard, and blank) should be provided.
Process validation: Brazil and Paraguay require the process validation report if section 3.2.P.3.5, Process Validation and/or Evaluation, does not provide the summary of the validation report, including acceptance criteria results and conclusions.
Requirements Beyond ICH
ICH’s mission is to achieve greater harmonization worldwide to ensure safe, effective, and high-quality medicines are developed and registered. Harmonization is achieved through the development of ICH guidelines via a process of scientific consensus with regulatory and industry experts working side-by-side. Key to the success of this process is the commitment of the ICH regulators to implement the final guidelines.14
As described previously, ANVISA in Brazil and COFEPRIS in Mexico (recent member) are the only ICH members in the region, thereby, the perception described next will focus on ANVISA’s adherence to ICH requirements. Although ANVISA has been an ICH member since 2016, the implementation plan of ICH guidelines is still ongoing. It is expected that some local regulations beyond ICH will continue to exist. It is well-understood that the process of updating or creating regulations takes time and effort and that it must be done while juggling other priorities of reviewing and approving new drug applications and, more recently, fighting a serious health pandemic.
ANVISA’s analytical method validation15 and forced degradation1 requirements are examples of those local regulations that bring additional technical requirements and make a Brazil dossier different from a US or EU dossier (both ICH member countries) and drive the divergence between harmonized global registration process.
In-Country Testing of Imported Medicines
The requirement for in-country testing for imported medicines has been in place in several Latin American markets for a while. Argentina, Brazil, Chile, Mexico, and Uruguay require in-country testing and perform the analytical method transfer or validation locally, which means that dossier submissions in these countries are sometime delayed due to logistics, something that was heavily experienced during the COVID-19 pandemic when there were extreme challenges to shipping samples, reference standards, and reagents to conduct transfer/validation exercises locally. In addition, a method transfer protocol and report also need to be generated and included in the dossier, as appropriate.16
In Brazil, partial quality control tests are allowed when certain criteria are met including, but not limited to, the number of batches imported or the temperature and humidity monitoring and recording during transportation.17 Companies often do not request the waiver of the quality control testing because it is difficult and more costly to execute these measures.
In Chile, it is required to include specific tests in the local drug product specification, depending on the dosage form.18 The local specification must be followed for the testing and release process in the Chilean market. Among the additional tests required are in-process control tests and primary packaging type and material. Although there is no technical basis to perform in-process control tests in the finished drug product, the Chile regulatory agency, ISP, has not been accepting the scientific justifications to waive those additional tests required by the local regulation.
In general, in-country testing of imported medicines causes delays for the batch release, reduces the remaining shelf life on the product, and delays patient access to medicine while increasing the risks for potential drug shortages. It is a misuse of resources that also negatively impacts the environment.19
Considering that drug manufacturers have appropriate controls throughout the production process and supply chain to ensure product quality in line with recognized good manufacturing and distribution practices, the regulatory agencies should consider waiving the import testing requirement and focus on establishing risk-based approaches that are commensurate with the level of risks in accordance with ICH Q9 Quality Risk Management.20
Regulatory Flexibilities during the COVID-19 Pandemic
Like many regulatory agencies around the world, the Latin American health authorities implemented several regulatory flexibilities during the COVID-19 pandemic to (a) increase manufacturing capacity for COVID-19-related medicines, (b) accept globally generated documents in lieu of local requirements, and (c) rely on (full or partial) assessment reports from other regulatory agencies, with the aim of enabling more efficiency and agility in the approval/authorization of medicines and without compromising regulatory standards, patient safety, and product quality.
Another valuable trend that emerged from the COVID-19 pandemic was the acceptance of electronically generated regulatory documents, including CPPs. At the break of the COVID-19 pandemic, a backlog of issuing CPPs was observed. The European Medicines Agency (EMA) responded to this backlog by implementing a new system to issue electronic certificates of medicinal products. As of 30 March 2020, EMA no longer provides printed certificates: only electronically signed and authenticated certificates will be issued. The WHO endorsed the EMA’s decision and encouraged the various boards of health to accept this approach.21 Similarly, the FDA Center for Drug Evaluation and Research (CDER) began issuing electronic CPPs (eCPP) starting December 2021.22
This was positively received by all countries in Latin America and expanded to other types of documents where an expected “original” document or a document with wet signature would be required.
On a positive note, there are ongoing discussions at the WHO on the revision of its scheme and the value of the CPP in national regulatory authorities, with the aim of adopting more unified and efficient regulatory approaches/processes. A series of modifications have occurred to respond to the changing regulatory environment where the recommendation is that the CPPs should not be requested in countries that have the capability to conduct full quality, safety, and efficacy reviews unless they have a rationale to request one.9
The authors’ opinions are that some of the flexibilities allowed by the Latin American regulators should continue when the impact of the pandemic decreases, such as acceptance of electronically signed documents, flexibility on importing testing, either allowing for late submissions of the local analytical validation data or waiving the requirement, acceptance of risk-based approaches to streamline CMC packages and/or to justify the absence of local requirements, and ultimately the national regulatory authorities following ICH guidelines.
The key message spread during the pandemic is that the industries and regulators should apply the learnings from this unprecedented global experience to streamline their current processes and seek opportunities for greater international collaboration, either through reliance procedures and/or collaborative review initiatives such as the International Coalition of Medicines Regulatory Authorities (ICMRA).23
Conclusion
The global pandemic has demonstrated that applying regional solutions is inadequate and inefficient. More than ever, we need to work toward a global solution, prioritize the harmonization of technical requirements, and eliminate the ones that do not add value to product quality or safety but instead delay product availability in the region. Such a harmonized approach via the adoption of ICH should be considered as a foundation for a global process.
The positive learnings from the transforming experience of COVID-19 pandemic should be applied to the regulatory processes in the Latin American region moving forward. Risk-based approaches to CMC data and to local specific requirements, with patient-centric focus should be permitted, to ensure the timely supply of life-saving medicine to the patients in the region.
About the Authors



