Risk Assessment
The risk assessment steps may be considered the most important aspects of an overall QRM process. If the risks are not identified, analyzed, and evaluated properly, decisions about how to control risk cannot be made efficiently.
Part of the risk assessment process is to consider whether there are any possible pathways that might realistically give rise to formation of impurities. At the conclusion of the risk assessment, risk should be expressed in quantitative or qualitative terms, depending on the data available from the three steps of the risk assessment: risk identification, risk analysis, and risk evaluation.
Risk Identification
Risk identification, which starts with qualitative hazard identification, is the process by which existing hazards are identified, initially without assigning magnitude. All available data should be reviewed to address the question “What could go wrong?”
This first step in the risk assessment process also identifies possible consequences. It serves as a prelude to the later steps of risk analysis and risk evaluation that ultimately lead to the appropriate control and management of risk.
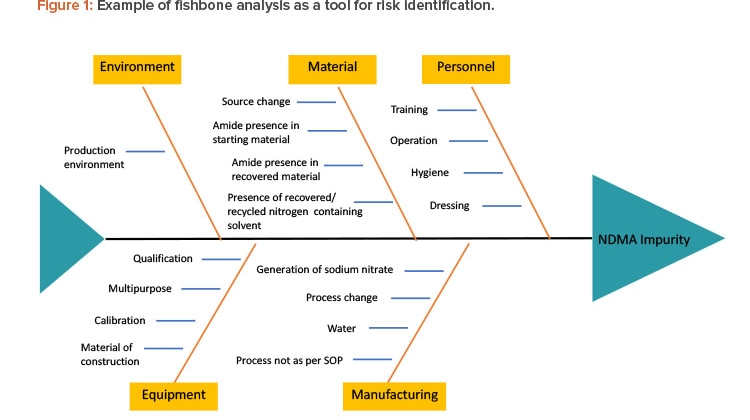
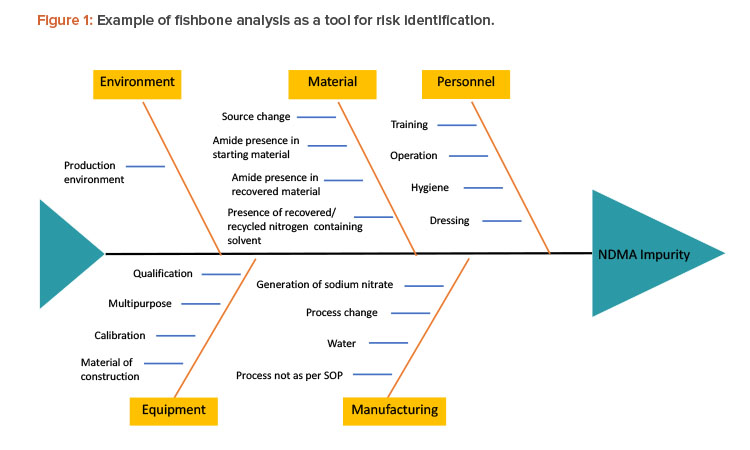
There are various tools to identify risk, such as fishbone diagrams, process mapping, and process breakdowns. To identify the risks of NDMA in the manufacturing process of a pharmaceutical product, one must understand the complete chemistry of NDMA—its chemical structure, chemical and physical properties, possible pathways by which it can be formed—as well as complete process mapping, complete details of ingredients used in manufacturing, interactions between different ingredients and environmental factors, and so on. Figure 1 presents a fishbone analysis identifying risk factors that may lead to the formation of impurities.
Risk Analysis and Evaluation
In these risk assessment steps, identified risks are analyzed and evaluated either qualitatively or quantitatively. Various tools are used worldwide to analyze and evaluate risk. Among these tools, failure mode effective analysis (FMEA), failure mode, effects, and criticality analysis (FMECA), fault tree analysis (FTA), process mapping, and hazard analysis and critical control point (HACCP) are used primarily in pharmaceutical sector., , , , , ,
Selecting the appropriate tool for risk assessment is of prime importance. For the purposes of NDMA risk assessment, FMEA is used here. It is a widely used tool for risk management of processes and can be useful to proactively identify failure modes, evaluate their impact, and determine process steps that must be changed.
FMEA includes a review of the following process steps:
- Failure modes (What could go wrong?)
- Failure causes (Why would the failure happen?)
- Failure effects (What would be the consequences of each failure?)
In FMEA, risk is scored on the basis of failure severity, probability, and detectability (see Tables 1–3)., Eventually, the FMEA conclusion is drawn on the basis of a risk priority number (RPN), which is a composite of scores for the three factors.
Table 1: Example FMEA severity criteria.
| Value |
Description |
Criteria |
| 1 |
Negligible |
No impact to product quality and process
robustness |
| 2 |
Marginal |
No impact to product quality |
| 3 |
Moderate |
Noticeable impact to product quality, but can be
recovered by reprocessing |
| 4 |
Critical |
Definite impact to product quality and patient safety |
| 5 |
Catastrophic |
Batch failure; not recoverable by rework; serious
concern for patient safety |
Table 2: FMEA probability of failure mode criteria.
| Value |
Description |
Criteria |
| 1 |
Rare |
Failure every 10–30 years |
| 2 |
Unlikely |
Failure every 5–10 years |
| 3 |
Possible |
Failure every 1–5 years |
| 4 |
Likely |
Failure more than once a year |
| 5 |
Almost certain |
Failure several times a year |
Table 3: FMEA detectability of failure mode criteria.
| Value |
Description |
Criteria |
| 1 |
High degree of
detectability |
Validated automatic detection system that is
a direct measure of failure, direct or indirect
indication of failure (e.g., control range and in
process control) |
| 2 |
Likely to detect |
Nonvalidated (manual or automated) detection
(e.g., visual level check, visual inspection of
vessels) |
| 3 |
Low or no
detectability |
No ability to detect the failure |
The probability criteria assess the frequency (rate of occurrence) of a given failure mode., Table 2 applies a linear scoring scale to the probability of occurrence of failure modes associated with the manufacturing process.
In FMEA, detectability criteria are used to assess the likelihood that failure modes or their impact will be detected. Table 3 shows a linear scoring method used for these criteria in a manufacturing process of pharmaceutical product.
As noted previously, conclusions drawn from the FMEA are based on the RPN, composite score of the three failure mode criteria, which is calculated as follows:
RPN = (S × P × D)
where S is the severity criteria score; P, the probability of occurrence criteria score; and D, the detectability of occurrence criteria score.
The RPN scores categorize risk in three levels:
- Low: 1–10
- Medium: 11–20
- High: 21–75
Table 4: FMEA assessment of NDMA formation risk for one pharmaceutical product.
| Item |
Factor |
Failure Mode |
Existing Control |
Severity (S) |
Probability (P) |
Detectability (D) |
RPN = (P x S x D) |
Recommended Action for Risk Mitigation |
Probability (P2) |
Severity (S2) |
Detectability (D2) |
RPN2=(P2xS2xD2) |
| Human |
Operation |
Operation does not follow standard operating procedure (SOP), which may lead to generation of impurities |
Every operator involved in manufacturing is trained; second person is available to review critical operation |
2 |
1 |
2 |
2 |
|
|
|
|
|
| Hygiene |
Personnel involved in production are unhygienic, which may lead to cross contamination |
All personnel involved in manufacturing are trained and maintain good hygiene |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Gowning |
Dirty/used clothing may lead to cross contamination |
As per SOP, operators are given fresh pair of coveralls at start of shift |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Machine |
Equipment qualification |
Nonqualified equipment or failure in qualification status |
Equipment is qualified initially and requalified on defined intervals per the validation master plan |
2 |
1 |
1 |
2 |
|
|
|
|
|
| Material |
Material of construction is not of required pharmaceutical grade, which may lead to cross contamination |
Material of construction for all equipment is of required pharmaceutical grade |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Material |
Amide or amine |
Presence in any starting material |
Amide or amine is not present in any starting material |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Presence in intermediates |
Because amide or amine is not present in any starting material, there is no possibility of generation in recovered substance |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Source |
Change of material source |
Supplier qualification process in place prohibits any material to be the part of process until its qualification is done |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Water |
Presence of impurities in water |
Water is tested for NDMA using a validated method |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Method |
Change in manufacturing process |
Change in process may trigger NDMA impurity |
When change occurs, all methods and steps adopted for manufacturing are validated |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Manufacturing process |
In the manufacturing process, dimethylamine and nitrite may generate nitrosamine |
Because no starting materials or intermediates involved in the manufacturing process can generate dimethylamine and nitrites, nitrosamine impurities cannot be generated in manufacturing process |
5 |
1 |
1 |
5 |
|
|
|
|
|
| Environment |
Production environment pollution |
Cross-contamination on the production floor may generate nitrosamine impurity |
All possible pathways of cross contamination are controlled; e.g., dust generation is controlled via a closed process |
5 |
1 |
1 |
5 |
|
|
|
|
|
NDMA Case Study
A risk assessment study is conducted identify and devise a recommended corrective action to minimize risk of NDMA impurity. For this purpose, the failure modes most likely to cause the generation of NDMA impurities are identified and their risks given the current controls in the existing manufacturing process are assessed. Table 4 is an example of FMEA risk assessment after initial risk identification, which can subsequently be extended after recommended risk mitigation is implemented.
Risk Control
Risk control is a decision-making activity to either accept or reduce risk. Effort put forth to control the risk should be proportional to the risk’s significance and its outputs. During risk control, the following are addressed:
- Is the risk above or below the acceptable level?
- What will be done to reduce or eliminate the risk?
- What is the appropriate balance of benefits, risks, and resources?
Before evaluating the probability that a hazard could occur, or assessing the ability to detect whether the fault can be found before the harm occurs, existing controls should be identified. This helps assess the current situation with respect to risk severity. This step can also identify the level of effort required to eliminate or mitigate that risk.
Risk Review and Communication
Risk review and communication are an ongoing part of a quality management system. Participants in risk management should review outputs and results while taking into account new knowledge and experience. Once the risk management process is initiated, risk review will be an ongoing process to consider events affected by the earlier decisions made during the QRM process. Risk review includes reconsidering earlier risk acceptance decisions.
The risk analysis presented in Table 4 did not identify any risk in the process that may have led to the formation of NDMA impurities in one product. However, companies should take a comprehensive approach to risk assessment to ensure that there is no risk of impurity being generated in other pharmaceutical products. It is necessary for every pharmaceutical manufacturer to conduct a thorough risk assessment study related to the generation of impurities and put controls in place to ensure the patient safety.
Conclusion
Unwanted impurities are among the biggest challenges facing pharmaceutical manufacturers and regulators. These impurities can pose a serious threat to the health of patients. The recent NDMA alerts for angiotensin receptor antagonists (valsartan, ranitidine, and metformin) have fueled the issue. In response, pharmaceutical manufacturers have taken various steps, such as product recalls and alerts, stringent supplier qualification programs, and routine batch testing for NDMA. Still, these efforts pale in comparison to the seriousness of issue.
In this regard, the strategy presented in this article to assess the control of impurities in the manufacturing process of pharmaceutical products via QRM may be crucial. When we apply QRM holistically to the manufacturing process, it will help us identify and devise control strategies for potential failure modes or gaps that could lead to the formation of impurities in product.