Technical
July / August 2021
Medical Device UDI Components Management in the European Union
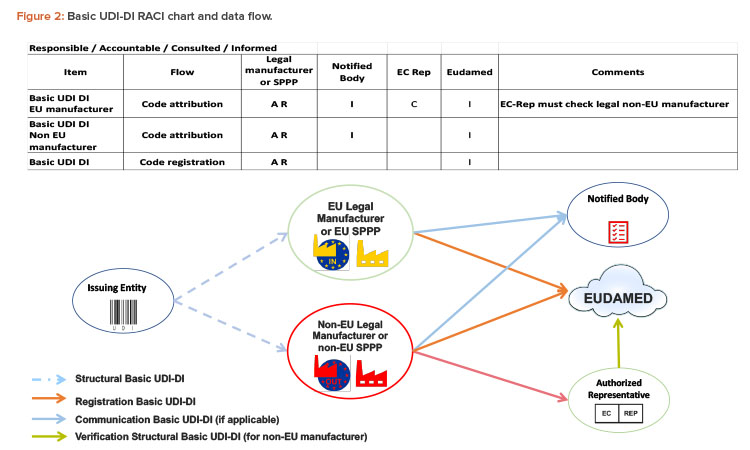
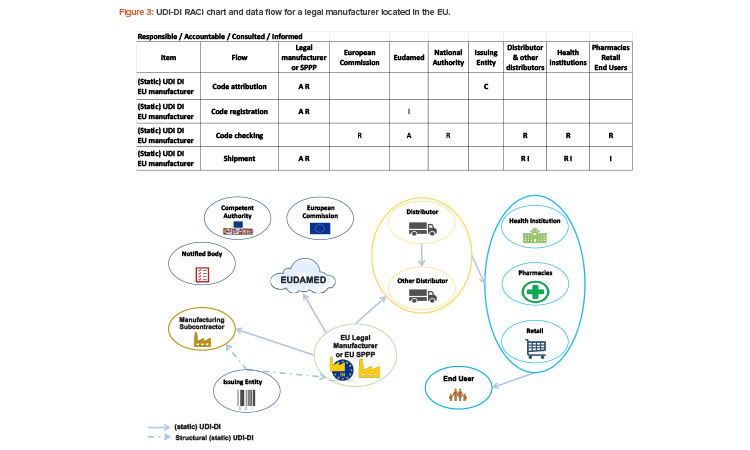
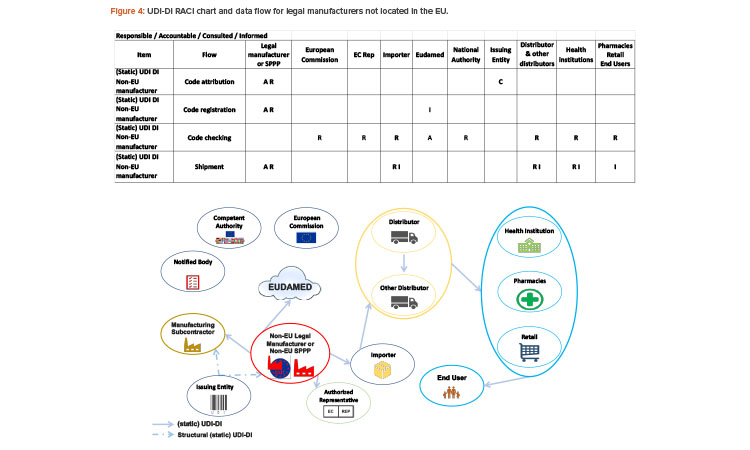
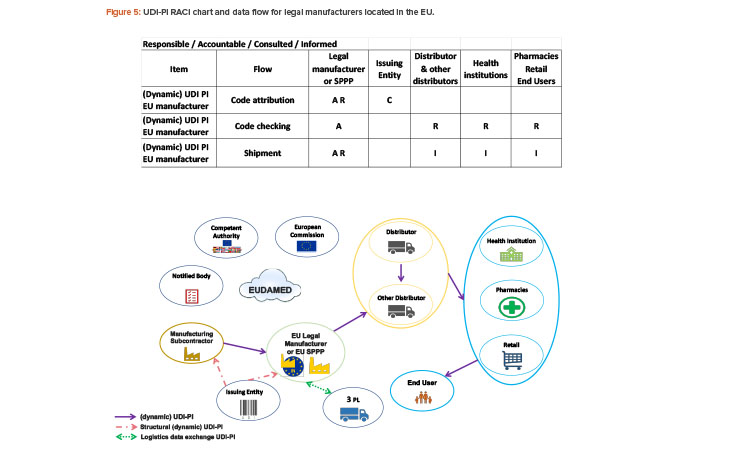
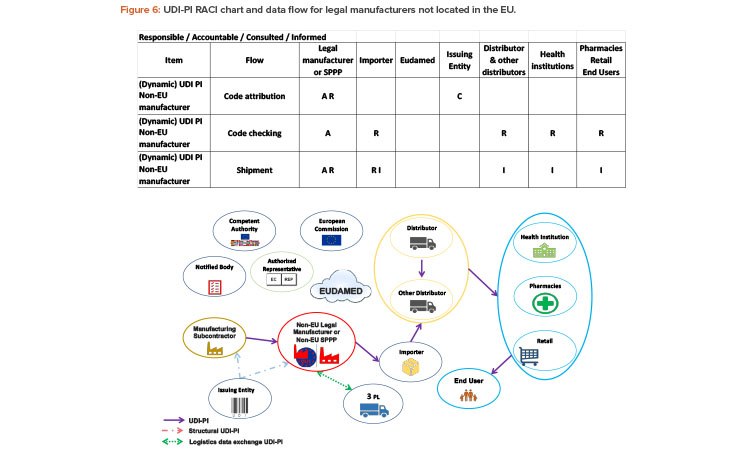
Since 2019, the ISPE France Affiliate’s Unique Device Identification (UDI) Medical Device Work Group has been producing tools to help project stakeholders within the EU or overseas understand and comply with EU regulations of UDIs in medical devices. Some of those tools are highlighted in the article.
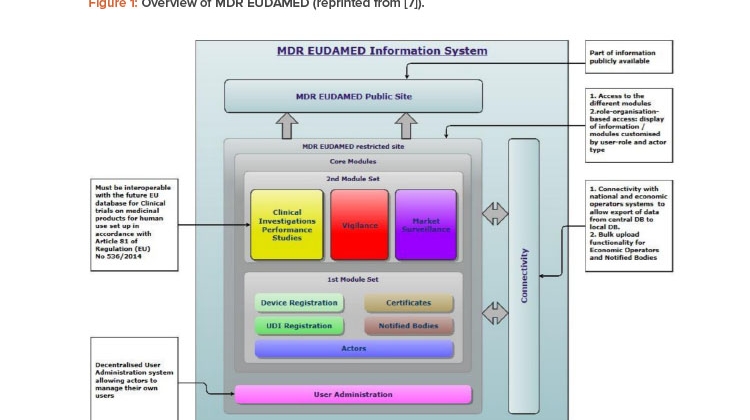
![Figure 1: Overview of MDR EUDAMED (reprinted from [7]).](/sites/default/files/2021-07/0721_PE_JA_TechUDI_01_0.jpg)
About the Authors

Laurence Azoulay has a masters in finance and marketing, and 26 years in the dental industry as Chief Purchasing Officer for a leading European dental...
Marie Coulon, PharmD, is a Regulatory Affairs Project Manager at Provepharm SAS, a molecule vitalization company in Marseille, France. After the completion of her pharmaceutical...
Christophe Devins is CEO of ACKOMAS. He worked with Gencod, which has since become the established GS1, after a master II degree. He is a...
Bernard Durand is a Computer Sciences Engineer in an industrial environment as project manager and business developer. He has worked in electronic goods production, glass...
Etienne Granier is a Packaging Expert at Laboratories Théa. Etienne sets up serialization of drug projects and launches UDI/EUDAMED projects.
Amel Guerrida Marchand is an IT Validation Specialist. Amel has been an ISPE member since 2019.
Ye-Lynne Lee is a Consultant in quality, regulatory, and vigilance of medical devices. She completed her pharmacy degree and graduated with a masters in medical...
Valerie Marchand has a masters degree in marketing and European economy, and has worked for more than 20 years in the healthcare sector at GS1...
Patrick Mazaud has been a Hospital Pharmacist for 30 years. He is a member of EuroPharmat, Scientific Society for Medical Devices in France, Counselor of...
Brigitte Naftalin holds a bachelors from the University of Mannheim and two masters in international business, commercial law, and languages from Universities of Freiburg, Germany...

Michel Raschas is an e-compliance expert and Consulting Validation & Services Director of PROGMP, a consulting, engineering, and validation company. He has an engineer degree...
Nadim Wardé is a Data Management Consultant focused on the product domain. With his broad experience, he supports companies in their product data journey through...


