The Application of GAMP® 5 to the Implementation and Operation of a GxP Compliant Clinical System

The need to validate computerized systems used in the context of a clinical trial has increasingly become the focus of regulatory oversight as industry has changed from mainly paper-based processes to electronically supported processes, particularly in the last decade.
1 Introduction
The principles and processes for the validation of computerized systems are well-known and understood within the Good Manufacturing Practice (GMP) and Good Laboratory Practice (GLP) sectors of the life science industry, and the GAMP® Guide, now in its fifth revision, is well-established as a recognized industry guideline for these processes. The origin of the GAMP® 5 Guide is based upon concepts developed over 20+ years within the pharmaceutical manufacturing sector, but during this time the fundamental principles of the risk-based life cycle approach have been expanded to be equally applicable across all GxP regulated industry areas. However, the guideline does not cover those requirements specifically applicable to the field of Good Clinical Practice (GCP). The need to validate computerized systems used in the context of a clinical trial has increasingly become the focus of regulatory oversight as industry has changed from mainly paper-based processes to electronically supported processes, particularly in the last decade.
The fundamental principles and concepts are identical whether computerized systems are operated in GMP and GCP environments, however there is a need to identify the relevant similarities and differences and to define some best practices for applying GAMP® 5 principles in the context of GCP systems. In this concept paper, we will demonstrate how the GAMP® 5 principles can be applied to the validation of a key system used in clinical trials, an Electronic Data Capture (EDC) system, and explore particular aspects to be considered when implementing these systems.
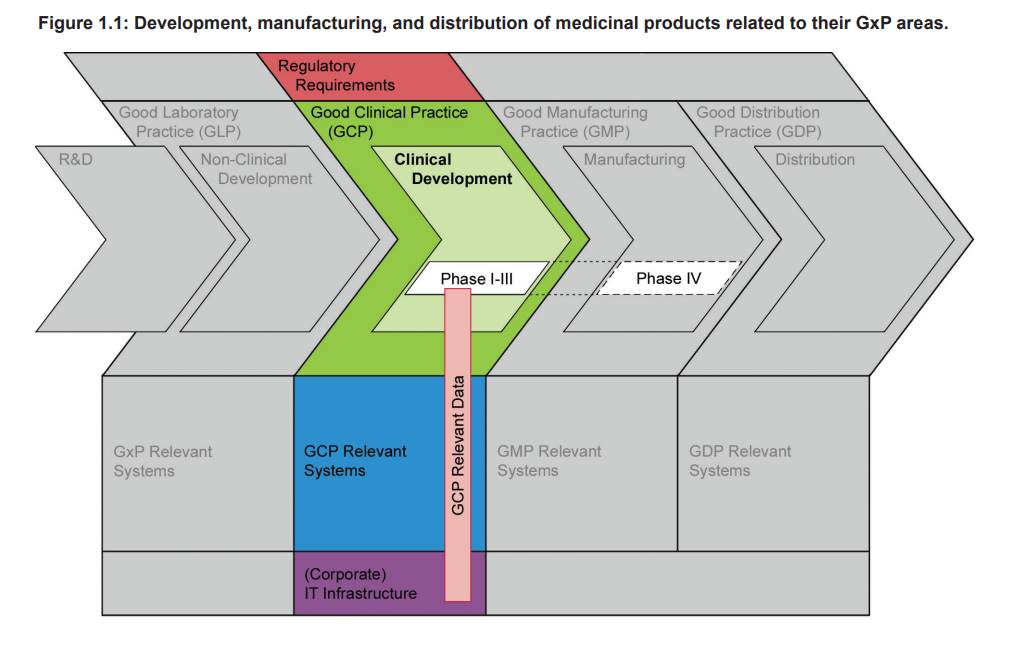
Figure 1.1 highlights that Clinical Development of a medicinal product is an essential part of the whole product life cycle, and one that has to be conducted in compliance with GCP. The diagram shows the phases of development, manufacturing, and distribution of medicinal products related to their GxP areas, and that there is Guidance gap between GMP and GLP.
2 Similarities and Differences between GMP and GCP Systems
2.1 Product Quality vs. Data Integrity
The GAMP® 5 Guide states that “patient safety is affected by the integrity of critical records, data, and decisions, as well as those aspects affecting physical attributes of the product” and product quality in the GMP environment is defined as the quality of a manufactured physical product that is directly consumed by or used on patients. Product quality is therefore of paramount importance as people might be harmed by products of poor quality. Companies manufacturing medicinal products are very aware of the quality requirements and the regulatory controls. They address these requirements through the implementation of a Quality Management System – an organizational structure and implementation of formally documented procedures, processes, and activities (e.g., extensive training) often supported by processes for the thorough testing and validation of computerized systems.
Ensuring the integrity of data managed by a computerized system is essential to support the evaluation of product quality and ultimately protect patient safety. The result of a clinical trial is not a physical product but rather data relating to the safety and efficacy of an Investigational Medicinal Product (IMP); always assuming that the IMP is of sufficient product quality. The quality parameters that can be evaluated for a clinical trial are correctness of the data and the constantly maintained integrity of data throughout the complete clinical trial. As data are collected, analyzed, processed, and corrected continuously, data integrity is at risk throughout the entire lifetime of the study. Since all data collected originate from study subjects, the protection of personal data and patient confidentially provide additional challenges that are more predominant than in most GMP environments.
2.2 Project Character of Trials
Every clinical trial is conducted and managed as an independent project even if they use the same IMP. Each clinical trial may be different as each addresses different parts of the development cycle (Phase I to IV or Non-Interventional Studies [NIS]) or varying product indications or endpoints. Trial projects, especially across the various phases, vary greatly in terms of duration, number of patients to be recruited, the pace of enrollment, and the spread of geographic location(s) involved.
The life cycle of a GCP system configuration, or in some cases the system itself, is primarily limited to the duration of the trial, while GMP systems are often in place for the lifetime of the manufactured product. While few trials run longer than three years, marketed products often exist for decades. Even though GCP systems are often used for multiple studies (e.g., Electronic Data Capture (EDC) system platforms), these systems must be adapted, configured, and sometimes deployed for each study separately to meet the trial specific requirements.
There are time pressures in the project/trial setup phase because the more time required for the development of a product, the less time that product can be marketed exclusively under a patent. Moreover, each trial investigates a different product or product aspect with a unique set of data to be collected and processed.
2.3 Control Over Technology, Process, and Training
In general, a validated computerized system is based on qualified infrastructure, validated software, qualified (site) personnel, and well-defined processes. But for a number of GCP systems, for example, EDCs, web portals, and Interactive Voice Response Systems/Interactive Web Response Systems (IVRS/IWRS), the scope of those system elements that are under the complete control of the system owner and/or users is limited. However, it is recognized that the infrastructure and the personnel of the organization involved in conducting the trial (Sponsor and/or Contract Research Organization [CRO]) can be as well-controlled as within a GMP environment. But GCP systems are often accessed by hundreds or thousands of users at the clinical investigator sites included in the trial, e.g., hospitals or general practitioners offices. These are typically outside of the sponsor organization, only bound by contracts and very often only participating in a few trials with the same sponsor. Therefore, the operation, maintenance, and control of the system must consider the diverse infrastructure and the differences in organization, experience, and training. Even if detailed contracts are in place, these investigational sites are usually not under the direct and/or permanent control of the process and system owner. For example, sponsor SOPs do not usually apply at the clinical site(s).
Therefore, the end user group that is likely to interact with a clinical trial system may be very diverse and will include physicians working in their own practice as well as investigators located in huge hospitals. In most cases, this results in systems that will be accessed via a local IT infrastructure that ranges from a semi-private laptop to a highly restricted hospital IT environment. Consequently, the sponsor has very limited control over this part of the IT infrastructure that is used in the clinical trial. Challenges here range from no control over the upgrade of the software/ operating system used locally, protection against malware etc., up to restrictions imposed by the local IT departments, e.g., with regard to available ports or specific firewall settings.
A simple example for this is browser software. Most investigator-facing clinical trial systems are web-based and accessed through web browsers. While it is simple to control the browser software within a company, site staff may use any browser in any available version with different combinations of plug-ins and add-ons (e.g., Java or Flash) on PCs, tablets, or even smartphones. Even limiting the allowed browser software by contractual agreement may not always be successful, as the investigator may be part of a larger organization (e.g., a large clinic) with its own standards and therefore might not be allowed to use the exact browser that has been specified and tested.
Consequently, a web-based investigator-facing system must be as independent from the software installed on the PC at the sites as possible. This is often referred to as Zero Footprint Applications (ZFAs, also called Zero Footprint Clients or Zero Footprint Software) referring to systems that do not require specific local resources or setup and do not require end users to install any software. The overall infrastructure consists of highly controlled components, e.g., the servers where the system is hosted (including physical as well as logical controls), and the lesser controlled components used for accessing the system by the sites.
But any patching of security vulnerabilities in the browser at the clinical site could potentially impact a web-based GCP system’s user interface and underlying system functionality. Recently, clinical trial systems, like EDCs, have also been interfaced with local systems such as Electronic Health Records (EHR). These may require further validation activities, which will not be described in more detail in this concept paper. The general question raised with these systems is: which controls are needed so that it is possible to import data from a non-validated system into a validated system and use it for GCP purposes?
Typically in very large trials, the training of the end users is frequently provided electronically and/or by a large team of people, potentially in multiple languages. Additionally, the users come from diverse backgrounds, such as outside of the sponsor/Contract Research Organization and are often not fully familiar with the technology. These aspects lead to varying degrees of comprehension and understanding within the user group from the training delivered and the potential for errors during the data entry process utilized for the EDC system.
This risk can be mitigated by configuring a user interface design and functionality that supports efficient and accurate data entry in the electronic Case Report Form (eCRF), and edit checks that directly identify, and therefore avoid, the most typical errors. The challenges in the area of training and qualification of site staff may lead to an increased need for support both for the specific trial and for the overall system. The responsible validation and technology teams should take this into account when planning the on-going support and maintenance of the system as well as the corresponding operational support processes.
In summary, the validation of GCP systems may need to address the following:
- System users may be located in a large number of different companies or organizations in different countries all over the world.
- System users may be not be following company Standard Operating Procedures (SOPs), corporate standards, or policies.
- Participating users may be restricted by local policies (e.g., IT security policies at hospitals).
- Locally used infrastructure may be very variable and complex (e.g., operating systems or browser).
- Changes in staff are not governed and controlled centrally.
Consequently, not all elements of the computerized system are as strictly controlled as they typically are in GMP systems. This severely influences the design of the system as it will need to:
- be as independent from the local infrastructure as possible
- use technical standards applicable across all sites
- address local staff changes
2.4 Raw Data vs. Source Data
In GMP systems, raw data are typically defined as any work sheets, (quality) records, memoranda, or notes that are the result of original observations, findings, measurements, or activities. Typically these data have not been manipulated or processed by other means. These data (or data derived from these) are the basis for GMP relevant decisions and activities. These raw data have not been subject to interpretation and have been collected in a tightly controlled process.
Source data in the GCP environment are “all information in original records and certified copies of original records of clinical findings, observations, or other activities in a clinical trial necessary for the reconstruction and evaluation of the trial” 4. Therefore, if source data are only electronic, specific regulatory requirements apply beyond FDA21 CFR Part 11.
The basis for GCP relevant decisions and activities are source data that are mostly recorded by human beings in source documents (e.g., patient charts, lab print-outs) or potentially (from a sponsor perspective) in uncontrolled systems like an EHR. These data are often manually entered from paper source documents into a validated computerized system (e.g., EDC) by humans and are then subject to source data verification by the sponsor or CRO through the monitoring process. The investigator is responsible for the completeness and accuracy of the source data as well as for the data entered into a system and releases these data entries through signing the individual eCRFs. Documents containing any source data are part of the documentation of the trial and must be filed and archived.
The quality and integrity of the source data is ensured by a system of mutual checks between the investigator and the sponsor. The investigator owns the source data and always retains the original under their control. The sponsor monitors the investigator for data quality and compliance to general GCP rules. There is typically a third party concerned with this process – the regulatory health authorities, who could perform inspections at both the sponsor and the investigator sites to verify that data handling activities comply with relevant regulations. Therefore, the term “source data” has a very specific meaning in the GCP area, which includes this system of mutual checks. Therein lies the fundamental difference with the term “raw data” as typically used in the GMP world, which are created under the sole control of the manufacturer.
Furthermore, most data management systems also allow for hybrid trials that are using direct data entry by the sites as well as data entry from paper CRFs by the Sponsor or CRO. However, all data have to be entered in the data management system to enable further evaluation. As outlined above, in more and more trials GCP systems are interfaced to local EHR systems. These EHR systems may themselves be interfaced with other local systems that record data from clinical trial subjects. The identification and verification of the source data may be challenging in such setups.
Therefore, the effort to manage and maintain the quality and integrity of the source data across all sites globally is often significantly higher than managing raw data generated locally at a manufacturing site.
2.5 Mid-trial Changes
Change and configuration management procedures describe all activities starting from the go-live of a system until retirement. The coverage of these change and configuration control activities are applicable to the computerized system including all hard- and software components as well as the functional and technical documentation. Whereas in the GMP world a change of an approved drug producing system and the operating software is commonly a project, changes to GCP systems are often far more numerous and part of the day-to-day activities within the project “clinical trial.”
These mid-trial changes are possible at any time during the trial due to protocol amendments and these might affect all parts of the trial and/or supporting systems and documentation. Often the decision-making period to change the study protocol is long, but once the sponsor has prepared the protocol amendment and this has been submitted to the relevant ethics committees and competent authorities, these regulatory bodies have to respond within clearly defined timelines. Within these timelines, all required technical changes, process changes, and training changes have to be assessed for potential impact and risk. These changes also need to be designed, prepared and, if required, validated to be ready for release into production. As these changes are applied when the trial has already started, the need to complete the change to the system(s) in a controlled and timely manner is highly important. But the change of the trial configuration and GCP system(s) design can be on such a basic level that the effort required to restructure the system(s) is almost equal to building a new system. The more fundamental the change required, the greater the risks to data quality and integrity. Therefore, as with GMP systems, good change/configuration control procedures are equally essential for GCP systems to ensure adequate documentation, review, and approval.
3 Application to the Life Cycle of an EDC System
3.1 The eCRF as Part of the EDC System
An EDC system is used in clinical trials to electronically collect all patient data that are within the scope of the trial protocol. These data are typically organized in patient visits or from patient charts and can cover a wide range of attributes, e.g., demographic data, concomitant diseases and medications, (Serious) Adverse Events ([S]AEs), laboratory analysis results, and data on executed procedures. The trial specific EDC setup is designed on the basis of a final trial specific Case Report Form (CRF) and contains the eCRF plus additional functionalities. The eCRFs are created/adapted for each trial project based on the trial protocol and can contain complex calculations and data verification checks. Furthermore, the eCRFs are often provided in multiple languages, including complex character sets, e.g., for Chinese or Japanese sites. To adequately support the business process, in particular the data collection in a clinical trial, the EDC system must provide the eCRFs to the participating sites of a trial. Therefore the EDC, as a computerized system, does consist of the EDC platform and the eCRFs. Figure 3.1 shows the different levels of systems and processes on which a clinical trial is based. The eCRF is part of the trial specific front end of the EDC system (blue level), which is based on a qualified infrastructure (purple level) and in use with trial specific defined processes (green level).
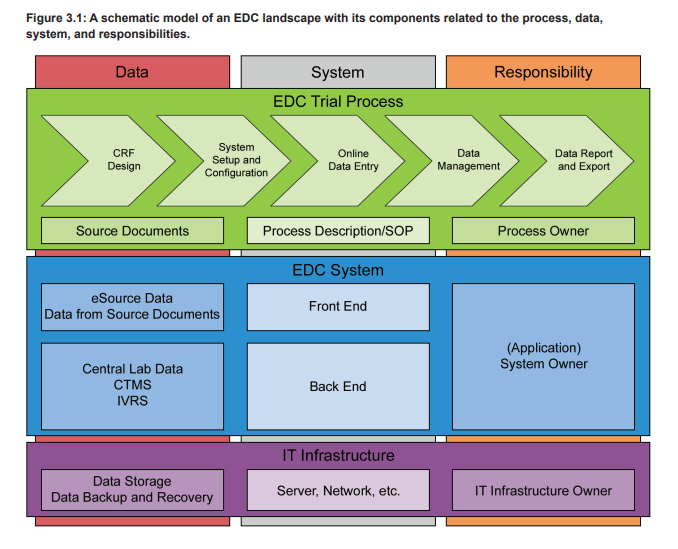
EDC systems are typically web-based and accessed by the local clinical trial teams via a web browser. The clinical trial teams enter the data after a patient visit into the provided eCRFs that perform real-time plausibility checks on the entered data. Additionally, the data are continuously cross-checked by the monitoring team of the sponsor or CRO. Any missing or incorrect data are followed up by the study team supported by manually initiated query workflows that are part of the EDC system. These workflows ensure reliable, efficient, and controlled communication between the clinical trial sites and the sponsor/CRO study team. As stated in ICH E6, the eCRF must be signed to document that the investigator or authorized member of the investigator’s staff confirms all observations recorded. If the CRFs are provided, completed, and signed electronically in an EDC system, the electronic signatures have to comply with FDA 21 CFR Part 11, EU GMP Annex 11, and other applicable regulations.
It is essential that the EDC system is stable and available at any time to ensure easy data-entry without delay, and to not limit the number of queries, especially if direct data entry by the sites is utilized. Furthermore, it is important to consider the expectations of the anticipated end users. Particular consideration should be given to the support of local languages and general ease of use, as otherwise the consistent use of the system and/or timely and correct entry of data may be jeopardized. Business continuity and disaster recovery processes are of special importance due to the huge number of continuously working end users at the sites. Another consideration is that the majority of modern EDC systems are often interfaced to other clinical systems, e.g., Clinical Trial Management System (CTMS) or Interactive Voice Response/Interactive Web Response (IVR/IWR) systems.
3.2 EDC System Setup – Platform Validation as the Foundation for eCRF Design
Modern EDC systems are typically designed and validated on a platform level, verifying and documenting that the required functionality to design and build eCRFs is available as expected. The study specific eCRFs utilize the available functionality and require a significant amount of configuration and/or customization, e.g., for edit checks to reduce the likelihood of incorrect or inconsistent data.
The trial specific configuration or customization required to build the necessary eCRFs need to be validated in the context of each trial. Recently, eCRF libraries have been implemented to increase the reusability of eCRFs or part thereof between trials to reduce the trial specific implementation effort.
Typically, EDC platforms are either complex Commercial Off-The-Shelf (COTS) systems (GAMP® category 4) or custom developed (GAMP® category 5). Based on system complexity it may be advisable to further classify additional elements of the system as GAMP® category 4 (e.g., configurable workflows that do not require programming).
A validated EDC platform provides the foundation for the subsequent eCRF design. However, as mentioned above, the eCRF design and the embedded plausibility checks do require extensive validation as well. So two separate life cycles, the platform life cycle and the study life cycle, must be considered when planning, validating, and operating an EDC system. When planning for validation, there should be a distinction made between the platform system (which typically has a development, test, and production server), and the trial specific applications, which often move through a development, test, and production environment on the production server of the platform system.
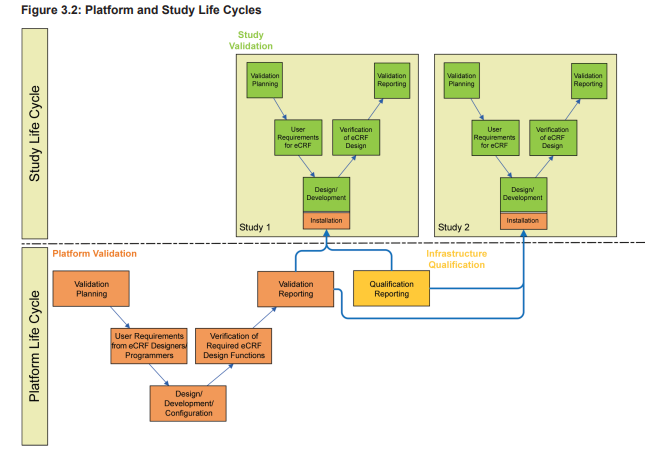
As illustrated in Figure 3.2, the platform life cycle is based on the requirements of the eCRF designers and programmers.
The study life cycle is based on the eCRF requirements of the clinical team supporting the study design including planned endpoints. As these life cycles may be independent from each other, at the planning stage the design of the system must consider the following question:
Can the EDC platform be updated or must it remain stable during the conduct of a trial?
| Strategy | Advantages | Disadvantages |
| Hold the EDC platform stable during the lifetime of the trial specific system | Reduce the risk to the eCRF, potential (study specific) interfaces and data integrity | Does not address IT security risks or technology advancements for the duration of the trial |
| Update the EDC platform during the lifetime of the trial specific system | Does allow installation of IT security patches or technology advancements | May lead to a significant number of deployments in different versions, therefore increases the IT support and maintenance efforts |
If the decision has been made to update the EDC platform, an impact analysis must be conducted to consider:
- Which features are changed, added, or removed?
- What approach to adopt for risk-based testing for all affected trial configurations?
It is important to document the results of the impact analysis and the testing approach to justify the upgrade decision.
Figure 3.3 show a potential upgrade scenario for an EDC platform leading to multiple EDC platforms supporting the ongoing studies. Such a scenario may increase the support effort significantly.
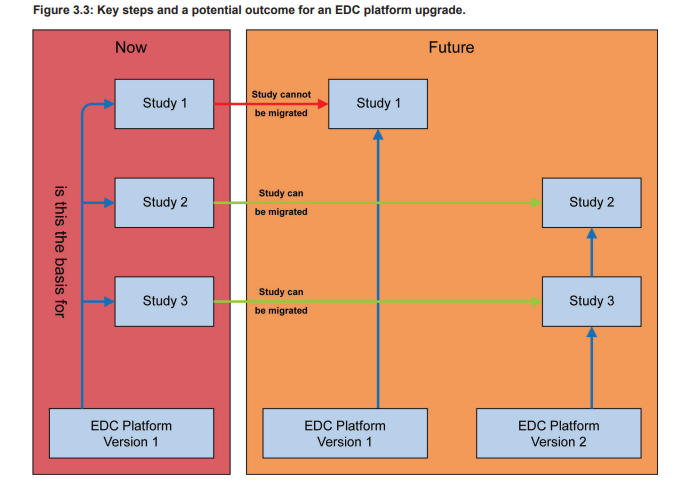
To minimize the support and maintenance efforts, the teams should aim to upgrade as many studies as possible. A first analysis of the platform upgrade should be irrespective of the individual affected trials, e.g., if the new version only provides bug fixes, it is very likely that they will not negatively impact any ongoing trials. However, if features are significantly changed, removed, or new features have been added, the analysis might need to consider study specific aspects in more detail. The decision to upgrade or to remain on an older version of the EDC platform must be well- justified (e.g., study is completed very soon) and documented.
The available COTS platforms are often hosted by the suppliers as Software as a Service (SaaS). If the EDC system is used as SaaS, parts of the validation are performed by the supplier and are often focused on the functional aspects including the functions for the eCRF design and the deployment/upgrade of the system. In this scenario it is essential for the sponsor to maintain a close cooperation with the system supplier to ensure that the validated state of the system is not endangered by uncontrolled changes.
3.3 eCRF Verification and Validation
Verification of the structure of the eCRF against the protocol is required to ensure that all required data are collected at all specified time-points needed for the appropriate statistical analysis required by the trial protocol. Ideally, all data are entered into the EDC system during the patient visit. However, current established practice is to enter data retrospectively into the EDC system after they have been recorded on source documents during the patient visit.
Validation of the EDC system is required to ensure that access is controlled, all information from each field is stored correctly in the database, and all calculations and data entry checks are accurate. Furthermore, since EDC systems store electronic records and typically use electronic signatures, compliance with Electronic Records/Electronic Signatures (ER/ES) regulations such as FDA 21 CFR part 11, EU GMP Annex 11, and other applicable regulations must be ensured. This includes verification that the audit trail is complete and reliable. All data including the data history and deleted data must be available in the database and displayed for review by the site/investigator/auditor/ inspector in the eCRF. For all “actions” the corresponding “timestamps” and the user identification must be recorded. This is stated in the Good Clinical Practice Guideline ICH E6 (Section 5.5.3 and 8.3.14/15) as well as Annex 11 of the EudraLex Vol. 4. GMP Guideline; where Annex 11 does not directly apply to GCP, but to GMP.
Verification of the data itself, by using automated checks on plausibility and monitoring of the data against the source data, is mandatory. Some checks cannot be automated as they involve judgments and require data management staff to look for complex relationships, for example, between disease states and concomitant medications.
3.4 System Design and Maintenance
System design and maintenance need to be considered during the risk assessments and design phase. Possible means to address the risks resulting from the lack of control at site mentioned above include, but are not limited to:
- Using a controlled IT-infrastructure (e.g., qualified server); same approach as for GMP systems
- Suitably qualified and trained people for the implementation process; same approach as for GMP systems
- Using a technology which can be used independently from the system on which the data are entered (e.g., web- based, using validation checks on the client and/or server side)
- Address fundamental system requirements (e.g., supported browser software and version) as part of the end user training
- Establish good lines of communication to end users for support with technology and other issues
- Have a well-organized support structure in place (e.g., according to ITIL®)
The validation team should be aware that testing of the EDC system cannot cover all possible scenarios. Therefore, particular attention should be taken during the system design phase to avoid imposing limitations with regard to browser software, underlying OS, Java, .net Framework, or even type of computer device used (e.g., tablets, smartphones) to access the EDC system. Such considerations should be documented in the formal risk assessment.
3.5 Change/Configuration Management
As mid-trial changes are likely to be the norm and not the exception, standardized and robust processes for change/ configuration management should be defined as part of the overall structure for the EDC/trial system life cycle. These processes need to be able to support a range of changes from a small change up to a complete eCRF redesign in the system. It is essential that the validation/technical team can assess the nature and impact of the change, document the resulting risks, and estimate the required effort quickly to support the planning of the mid-trial change. The implementation of the mid-trial change should not jeopardize the validated state of the overall system.
Unlike GMP, there is no clear regulatory requirement for Quality Assurance approval of changes to clinical computer systems before they go live, although the regulations do imply that such an action is required. As a matter of course, such actions should be performed as good practice. This emphasizes the need for effective change/configuration management procedures.
Summary and Conclusion
In summary, validation activities for GCP applicable systems must focus on the quality, security, and accessibility of the data and ensuring data integrity as the key quality parameters.
Essential Operational aspects should include:
Training of personnel on software life cycle processes and qualification steps for the EDC platform
OR
Qualification of the EDC platform supplier by the sponsor or delegate (e.g., CRO)
- Training of personnel on software life cycle processes and verification/validation steps for CRF/eCRF design
- Efficient and effective training of the end user
- System monitoring to ensure adequate on-site performance
- The establishment and management of appropriate support services (Maintenance Phase)
- Implementation of Operational Change and Configuration Management processes for EDC platform and study specific eCRF in parallel
As discussed above, the principles of GAMP® 5 can be applied to the validation of EDC systems. To further illustrate this point, the following table lists the terminology used in GAMP® 5 and the specific activities that may be required for a GCP critical system such as EDC.
| Traditional Term | GAMP® 5 Verification Activity | Possible EDC Validation Activity |
| Design Qualification (DQ) | Design Review | EDC platform Design Documentation
Trial Design Documentation
|
| Installation Qualification (IQ) | Checking, testing, or other verification to demonstrate correct:
| EDC platform Installation Documentation
Trial Installation Documentation
|
| Operational Qualification (OQ) | Testing or other verification of the system against specification to demonstrate correct operation of functionality | EDC platform Operational Documentation
Trial Operational Documentation
|
| Performance Qualification (PQ) | Testing or other verification of the system to demonstrate fitness for intended use | EDC platform Performance Documentation
Trial Performance Documentation
|
Major problem areas to be considered:
- The Supplier qualification and system evaluation process must be an integral part of the validation life cycle and must be maintained throughout the trial, especially in a SaaS type setup.
- Often very short timelines for trial setup and validation require an adequate routine validation framework for eCRF design and release.
- Issues specific to sites such as:
- the technology platform is not controlled by the sponsor or delegate, leading to problems that may not be completely resolved by the “Zero Footprint” approach
- the size, turnover, and diversity of the end user group lead to greater support and training needs
The following aspects are also important in the context of implementing an EDC system; however, they remain outside of the scope of this concept paper.
- Migration aspects to another EDC system (e.g., in case of bankruptcy of a supplier, or changes in business relationships)
- Cloud aspects of EDC systems including data hosting locations, data security, and privacy aspects
When comparing the risks and the resulting validation approach of GCP systems and GMP systems, using an EDC system as an example, it becomes obvious that the overall validation approach and basic principles as laid out in the GAMP® 5 Guide can be applied to both.
Crucially though, some aspects specific to GCP systems do require special consideration. For an investigator-facing EDC system, this includes the following aspects:
- The project-nature of clinical trials leading to overlapping but independent platform and study life cycles
- Less controlled system areas (e.g., infrastructure, staff qualification, segregation of duties) at the clinical trial sites
- Differences between raw and source data including local sources for source data
All of these aspects create unique risks as well as require individual validation approaches and/or the application of specific controls to mitigate.1,2,3
Abbreviations/Definitions
| COTS | Commercial off-the-shelf |
| CRO | Contract Research Organization |
| CTMS | Clinical Trial Management System |
| DQ | Design Qualification |
| eCRF | Electronic Case Report Form |
| EDC | Electronic Data Capture |
| EHR | Electronic Health Record |
| GAMP | Good Automated Manufacturing Practice |
| GCP | Good Clinical Practice |
| GLP | Good Laboratory Practice |
| GMP | Good Manufacturing Practice |
| GxP | A generic term to specify the underlying international pharmaceutical requirements, such as those set forth in the US FD&C Act, US PHS Act, FDA regulations, EU Directives, Japanese regulations, or other applicable national legislation or regulations under which a company operates. |
| IMP | Investigational Medicinal Product |
| IQ | Installation Qualification |
| IT | Information Technology |
| ITIL | Information Technology Infrastructure Library |
| IVRS | Interactive Voice Response System |
| IWRS | Interactive Web Response System |
| NIS | Non-Interventional Study |
| OQ | Operational Qualification |
| OS | Operating System |
| PC | Personal Computer |
| PQ | Performance Qualification |
| SaaS | Software as a Service |
| SAE | Serious Adverse Event |
| SOP | Standard Operating Procedure |
| UAT | User Acceptance Test |
| Zero Footprint | Computer applications which do not require end users to install any software (Also known as Zero Footprint Clients or Zero Footprint Software) |
Limitation of Liability
In no event shall ISPE or any of its affiliates, or the officers, directors, employees, members, or agents of each of them, be liable for any damages of any kind, including without limitation any special, incidental, indirect, or consequential damages, whether or not advised of the possibility of such damages, and on any theory of liability whatsoever, arising out of or in connection with the use of this information.
© Copyright ISPE 2013. All rights reserved.
All rights reserved. No part of this document may be reproduced or copied in any form or by any means – graphic, electronic, or mechanical, including photocopying, taping, or information storage and retrieval systems – without written permission of ISPE.
All trademarks used are acknowledged.
Acknowledgements
By the ISPE GAMP Community of Practice
This concept paper was written and reviewed by members of the Global ISPE R&D and Clinical Systems Special
Interest Group (SIG) of the ISPE GAMP Community of Practice (COP).
SIG Chairs
Oliver Herrmann Q-FINITY Quality Management Germany
Eric Staib ReSearch Pharmaceutical Services, Inc. USA
Document Authors
Marina Mangold Alcedis GmbH Germany
Frank Henrichmann PAREXEL International GmbH Germany
Oliver Herrmann Q-FINITY Quality Management Germany
Dieter Wachtmann PAREXEL International GmbH Germany
Reviewers
Aneta Blazevska Abbott GmbH & Co. KG Germany
Tamara Dehnhardt B.Braun Melsungen AG Germany
Ina Helm Alcedis GmbH Germany
Corinne Liabeuf Novartis Pharma AG Switzerland
Eric Staib ReSearch Pharmaceutical Services, Inc. USA
Maximilian Stroebe Novartis Vaccines and Diagnostics Netherlands
Frank Woods GlaxoSmithKline United Kingdom
Regulatory Input and Review
Christa Färber Trade and Industrial Agency of State of Germany
Lower Saxony – Agency Hannover Jonathan Helfgott CDER FDA USA
Particular thanks go to the following for their support of this Concept Paper: Chris Reid Integrity Solutions Ltd. United Kingdom
Michael Rutherford Eli Lilly and Company USA
Additional thanks to: Chris Clark Napp Pharmaceuticals Ltd. United Kingdom
Sion Wyn Conformity Ltd. United Kingdom


